BAUH_genotype-guided-donor-assignment
C.B. Azodi
7/29/2021
Last updated: 2021-08-06
Checks: 6 1
Knit directory: BAUH_2020_MND-single-cell/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 0822426. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Untracked files:
Untracked: .snakemake/
Untracked: 2021-04-27_pilot2_nCells-per-donor.pdf
Untracked: BAUH_2020_MND-single-cell.Rproj
Untracked: GRCh38_turboGFP-RFP_reference/
Untracked: Log.out
Untracked: Rplots.pdf
Untracked: analysis/2021-05-18_BAUH_STARsolo.Rmd
Untracked: analysis/2021-07-07_geno-guided-donor-assignment-tests.Rmd
Untracked: analysis/2021-07-14_genotype-kinship.Rmd
Untracked: analysis/2021-08-04_DropletQC.Rmd
Untracked: code/10x_2_h5ad.py
Untracked: code/dropkick_extract_cell_barcodes.py
Untracked: code/dropkick_get_ambient_and_hvgs.py
Untracked: code/function_vireo.R
Untracked: code/run_soupX.R
Untracked: code/test_dropletQC.R
Untracked: config/config_pilot2.1.yml
Untracked: star-help.txt
Untracked: test/
Untracked: test_learn/
Untracked: test_maf01_notFiltered/
Untracked: test_maf05/
Untracked: test_noGeno/
Untracked: test_vireo/
Untracked: workflow/config_ambient_pilot2.1.yml
Untracked: workflow/envs/ambient.yaml
Untracked: workflow/envs/dropkick.yaml
Untracked: workflow/snake_ambient.smk
Untracked: workflow/snake_ambient_vcfs.smk
Untracked: workflow/snake_non_ambient.smk
Unstaged changes:
Modified: analysis/index.Rmd
Modified: config/config_pilot2.starsolo.yml
Modified: workflow/Snakefile-single
Modified: workflow/rules/star-solo_workflow.smk
Modified: workflow/rules/vireo_single.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
suppressPackageStartupMessages({
library(corrplot)
library(tidyverse)
library(RColorBrewer)
library(ggpubr)
library(ggupset)
})
source("code/function_vireo.R")
d.sporadic <- c("81", "82", "84", "91", "98", "100", "106", "152", "154",
"184", "207", "231", "239")
d.control <- c("W001", "W104", "W164", "W220", "W221", "W222", "W263")
d.C9Orf72 <- c("114", "149")
d.SOD1 <- c("131")
d.TDP43 <- c("132")
d.cols.simple <- c("control" = "gray50", "sporadic" = "#66CCEE",
"SOD1" = "#CCBB44", "TDP43" = "#AA3377", "C9Orf72" = "#228833",
"doublet" = "#CC3311", "unassigned" = "#EE7733")
d.cols <- data.frame(list(donor=c(d.sporadic, d.control, d.C9Orf72, d.SOD1,
d.TDP43, "doublet", "unassigned"),
type = c(rep("sporadic", length(d.sporadic)),
rep("control", length(d.control)),
rep("C9Orf72", length(d.C9Orf72)),
rep("SOD1", length(d.SOD1)),
rep("TDP43", length(d.TDP43)),
"doublet", "unassigned")))
d.cols <- merge(d.cols, as.data.frame(d.cols.simple), by.x="type", by.y="row.names")Vireo results
Early tests
test.list <- c("test/",
"test_learn/",
"test_maf01_notFiltered/",
"test_maf05/",
"test_noGeno/")
test.id <- c("FilterAmb100_MAF0.01_fixed",
"FilterAmb100_MAF0.01_learnGT",
"FilterAmb100_MAF0.01_learnGT_notfiltered",
"FilterAmb100_MAF0.05_fixed",
"FilterAmb100_MAF0.01_denovo")
geno.res <- joinVireoResults(test.list, ids=test.id, key=d.cols)
geno.res.summary <- geno.res %>% group_by(id) %>%
filter(donor != "unassigned" & donor != "doublet") %>%
summarise_at(vars(n), list(min = min, median = median,
mean = ~round(mean(.),2), max = max)) %>%
arrange(desc(median))
geno.res.summary# A tibble: 5 x 5
id min median mean max
<chr> <dbl> <dbl> <dbl> <dbl>
1 FilterAmb100_MAF0.01_denovo 0 7.5 192. 4600
2 FilterAmb100_MAF0.01_fixed 0 0 0.3 2
3 FilterAmb100_MAF0.01_learnGT 0 0 237. 10912
4 FilterAmb100_MAF0.01_learnGT_notfiltered 0 0 0.13 2
5 FilterAmb100_MAF0.05_fixed 0 0 4.17 63Result: Very poor genotype assignment, need to look into this more.
Genotype guided assignment
loc <- "output/pilot2.1_gex/05_vireo"
geno.res.list <- c(paste0(loc, "/CB-scRNAv31-GEX-lib01_S1/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.01_no152/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.01_no154/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.02/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.03/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.04/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.05_no152/"),
paste0(loc, "/CB-scRNAv31-GEX-lib01_S1_MAF0.05_no154/"),
paste0(loc, "/test_CB-scRNAv31-GEX-lib01_S1/"),
# Filtering SNPs instead of the BAM
paste0(loc, "-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
# Filtering out ambient genes
#paste0(loc, "/amb100/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
#paste0(loc, "/amb100/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/ambient1000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/ambient1000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/ambient5000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/ambient5000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
# Filtering in HVGs genes
paste0(loc, "/hvgs1000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/hvgs1000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/hvgs5000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/hvgs5000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/hvgs10000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/hvgs10000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
# Filtering out ambient genes with ForceLearn
paste0(loc, "/learnGT_ambient1000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/learnGT_ambient1000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/learnGT_ambient5000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/learnGT_ambient5000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
# Filtering in HVGs genes with ForceLearn
paste0(loc, "/learnGT_hvgs1000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/learnGT_hvgs1000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.05/"),
paste0(loc, "/learnGT_hvgs5000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "/learnGT_hvgs5000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.05/")) #,
#paste0(loc, "/learnGT_hvgs10000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
#paste0(loc, "/learnGT_hvgs10000/learnGT_CB-scRNAv31-GEX-lib01_S1_MAF0.05/"))
# IDs: ID_FilterDetails_MAF_Donors
id.list <- c("FilterBAM_Amb100_MAF0.01_all_learnGT",
"FilterBAM_Amb100_MAF0.01_no152_learnGT",
"FilterBAM_Amb100_MAF0.01_no154_learnGT",
"FilterBAM_Amb100_MAF0.02_all_learnGT",
"FilterBAM_Amb100_MAF0.03_all_learnGT",
"FilterBAM_Amb100_MAF0.04_all_learnGT",
"FilterBAM_Amb100_MAF0.05_all_learnGT",
"FilterBAM_Amb100_MAF0.05_no152_learnGT",
"FilterBAM_Amb100_MAF0.05_no154_learnGT",
"FilterBAM_Amb100_MAF0.01_all_fixed",
# Filtering SNPs instead of the BAM
"FilterSNPs_HWE0.001_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001_MAF0.05_all_fixed",
# Filtering out ambient genes
#"FilterSNPs_HWE0.001-Amb100_MAF0.01_all_fixed",
#"FilterSNPs_HWE0.001-Amb100_MAF0.05_all_fixed",
"FilterSNPs_HWE0.001-Amb1k_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001-Amb1k_MAF0.05_all_fixed",
"FilterSNPs_HWE0.001-Amb5k_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001-Amb5k_MAF0.05_all_fixed",
# Filtering in HVGs genes
"FilterSNPs_HWE0.001-HVGs1k_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001-HVGs1k_MAF0.05_all_fixed",
"FilterSNPs_HWE0.001-HVGs5k_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001-HVGs5k_MAF0.05_all_fixed",
"FilterSNPs_HWE0.001-HVGs10k_MAF0.01_all_fixed",
"FilterSNPs_HWE0.001-HVGs10k_MAF0.05_all_fixed",
# Filtering out ambient genes with ForceLearn
"FilterSNPs_HWE0.001-Amb1k_MAF0.01_all_learnGT",
"FilterSNPs_HWE0.001-Amb1k_MAF0.05_all_learnGT",
"FilterSNPs_HWE0.001-Amb5k_MAF0.01_all_learnGT",
"FilterSNPs_HWE0.001-Amb5k_MAF0.05_all_learnGT",
# Filtering in HVGs genes with ForceLearn
"FilterSNPs_HWE0.001-HVGs1k_MAF0.01_all_learnGT",
"FilterSNPs_HWE0.001-HVGs1k_MAF0.05_all_learnGT",
"FilterSNPs_HWE0.001-HVGs5k_MAF0.01_all_learnGT",
"FilterSNPs_HWE0.001-HVGs5k_MAF0.05_all_learnGT") #,
#"FilterSNPs_HWE0.001-HVGs10k_MAF0.01_all_learnGT",
#"FilterSNPs_HWE0.001-HVGs10k_MAF0.05_all_learnGT")
geno.res <- joinVireoResults(geno.res.list, ids=id.list, key=d.cols)
geno.res.summary <- geno.res %>% group_by(id) %>%
filter(donor != "unassigned" & donor != "doublet") %>%
summarise_at(vars(n), list(min = min, median = median,
mean = ~round(mean(.),2), max = max)) %>%
arrange(desc(median))
geno.res.summary# A tibble: 30 x 5
id min median mean max
<chr> <dbl> <dbl> <dbl> <dbl>
1 FilterSNPs_HWE0.001-HVGs10k_MAF0.01_all_fixed 73 125 122. 191
2 FilterSNPs_HWE0.001-HVGs5k_MAF0.01_all_fixed 55 86.5 92.5 175
3 FilterSNPs_HWE0.001-HVGs10k_MAF0.05_all_fixed 37 81.5 80.4 160
4 FilterSNPs_HWE0.001-HVGs5k_MAF0.05_all_learnGT 31 56.5 70 241
5 FilterSNPs_HWE0.001-HVGs5k_MAF0.05_all_fixed 31 53 65.2 204
6 FilterSNPs_HWE0.001-HVGs1k_MAF0.01_all_fixed 11 39 39.5 108
7 FilterSNPs_HWE0.001-HVGs5k_MAF0.01_all_learnGT 9 23 217. 4611
8 FilterBAM_Amb100_MAF0.01_all_fixed 0 8.5 29.4 170
9 FilterSNPs_HWE0.001-HVGs1k_MAF0.05_all_fixed 1 8 9.96 30
10 FilterSNPs_HWE0.001-HVGs1k_MAF0.05_all_learnGT 2 7 9.92 44
# … with 20 more rowsPlot the number of cells assigned to each donor (and the median) for the best genotype-guided vireo runs:
top6 <- geno.res.summary$id[1:6]
geno.res %>%
filter(donor != "unassigned" & donor != "doublet" & id %in% top6) %>%
ggplot(aes(x=fct_reorder(id, n+1, .fun = median, .desc=FALSE), y=n+1, color=donor)) +
geom_jitter(width=0.2, alpha=0.5) +
coord_flip() + xlab("") +
scale_y_continuous(trans='log2') + theme_classic2(14) +
stat_summary(fun=median, geom="point", shape=18, size=3, color="black") 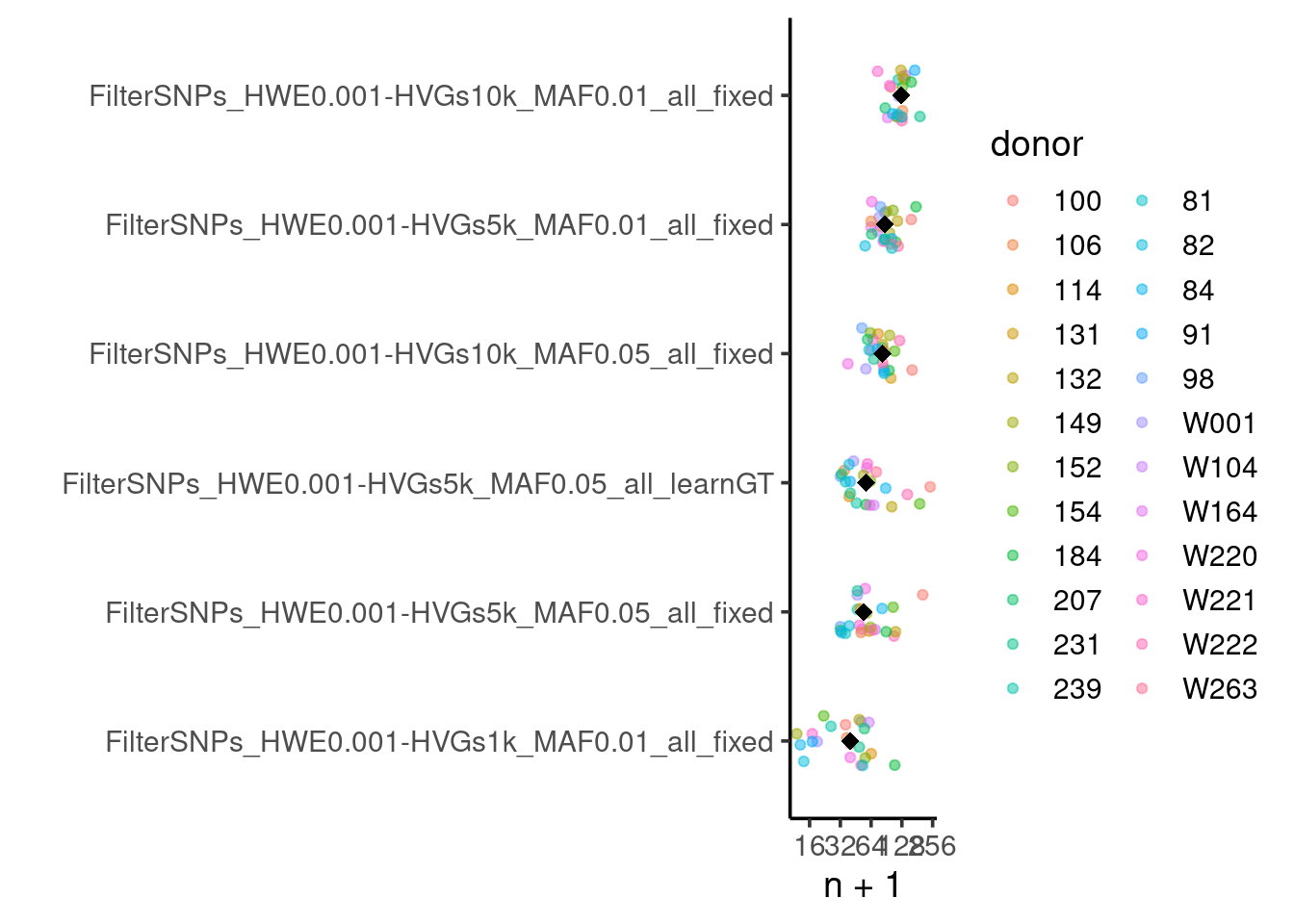
Consistency between vireo runs
Of the top performing models, do they consistently assign cells to the same donors? Table shows the number of cells with n donors assigned across the top 10 performing vireo donor assignments. Finding, most cells are only assigned to one donor, but plenty have conflicting assignments!
n <- 6
top <- geno.res.summary$id[1:n]
assignments <- joinVireoAssignments(geno.res.list, ids=id.list)
assignmentsTop <- assignments %>% select(all_of(top)) %>%
mutate(across(where(is.character), ~na_if(., "unassigned"))) %>%
mutate(across(where(is.character), ~na_if(., "doublet"))) %>%
filter(if_any(everything(), ~ !is.na(.)))
assignmentsTop$count <- apply(assignmentsTop[, top], 1, function(x) length(unique(x[!is.na(x)])))
assignmentsTop$present <- apply(assignmentsTop[,top], 1, function(x) unique(x[!is.na(x)]))
table(assignmentsTop$count)
1 2 3 4 5
4102 1504 275 24 1 Are there certain donors that are commonly confused? Finding:
length(unique(assignmentsTop[assignmentsTop$count > 1, "present"]))[1] 789assignmentsTop %>% filter(count > 1) %>%
ggplot(aes(x = present)) + geom_bar() + theme_bw() +
scale_x_upset(n_intersections = 20)Warning: Removed 1549 rows containing non-finite values (stat_count).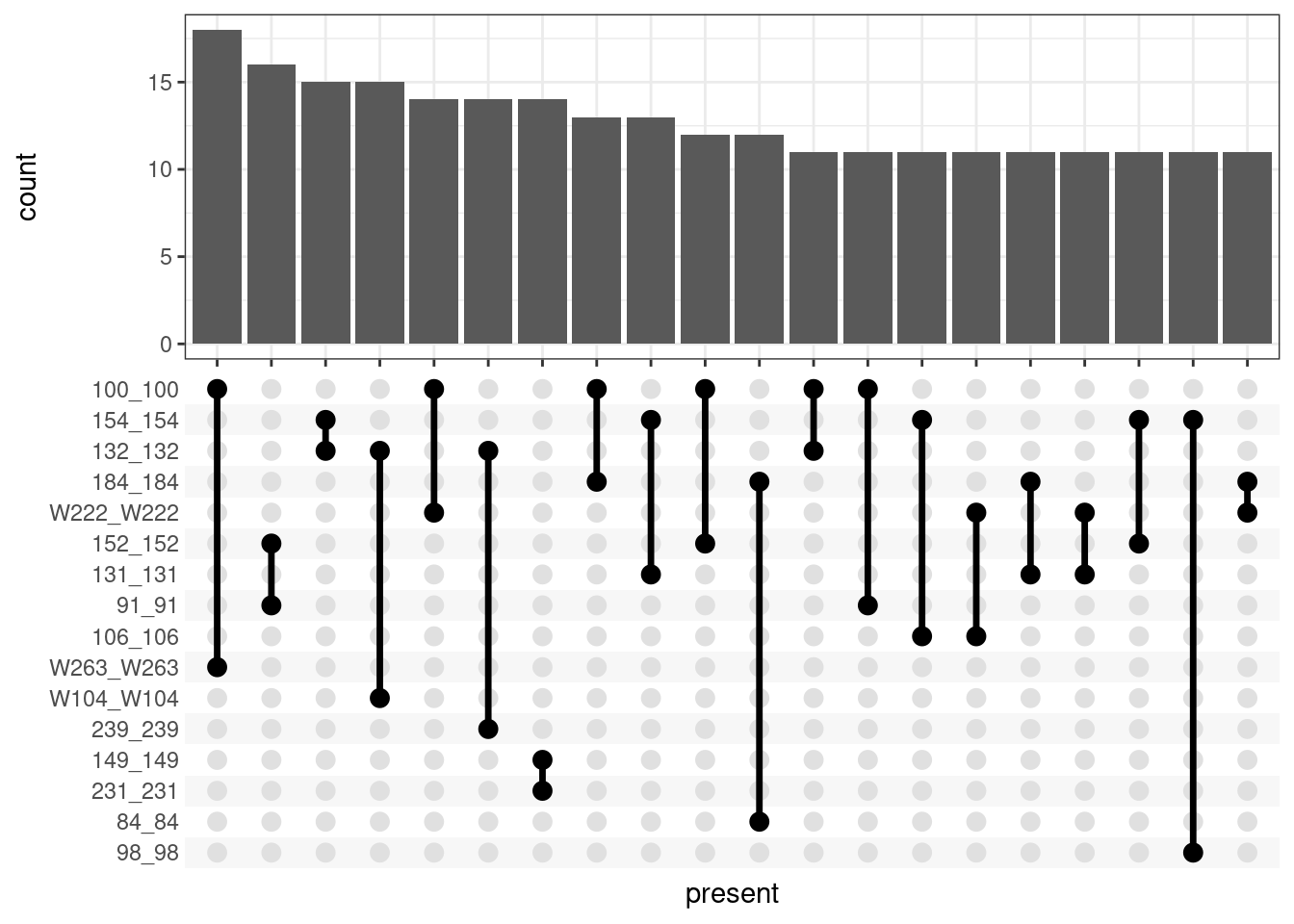
Focusing, just on the cells that don’t have conflicting results, how many of the top 10 models support the assignment?
assignmentsTop %>%
mutate(support = rowSums( !is.na( assignmentsTop[, top]))) %>%
filter(count == 1) %>% group_by(support) %>% summarise(n.cells = n()) %>%
ggplot(aes(x = as.factor(support), y = n.cells)) + geom_bar(stat="identity") + theme_classic2()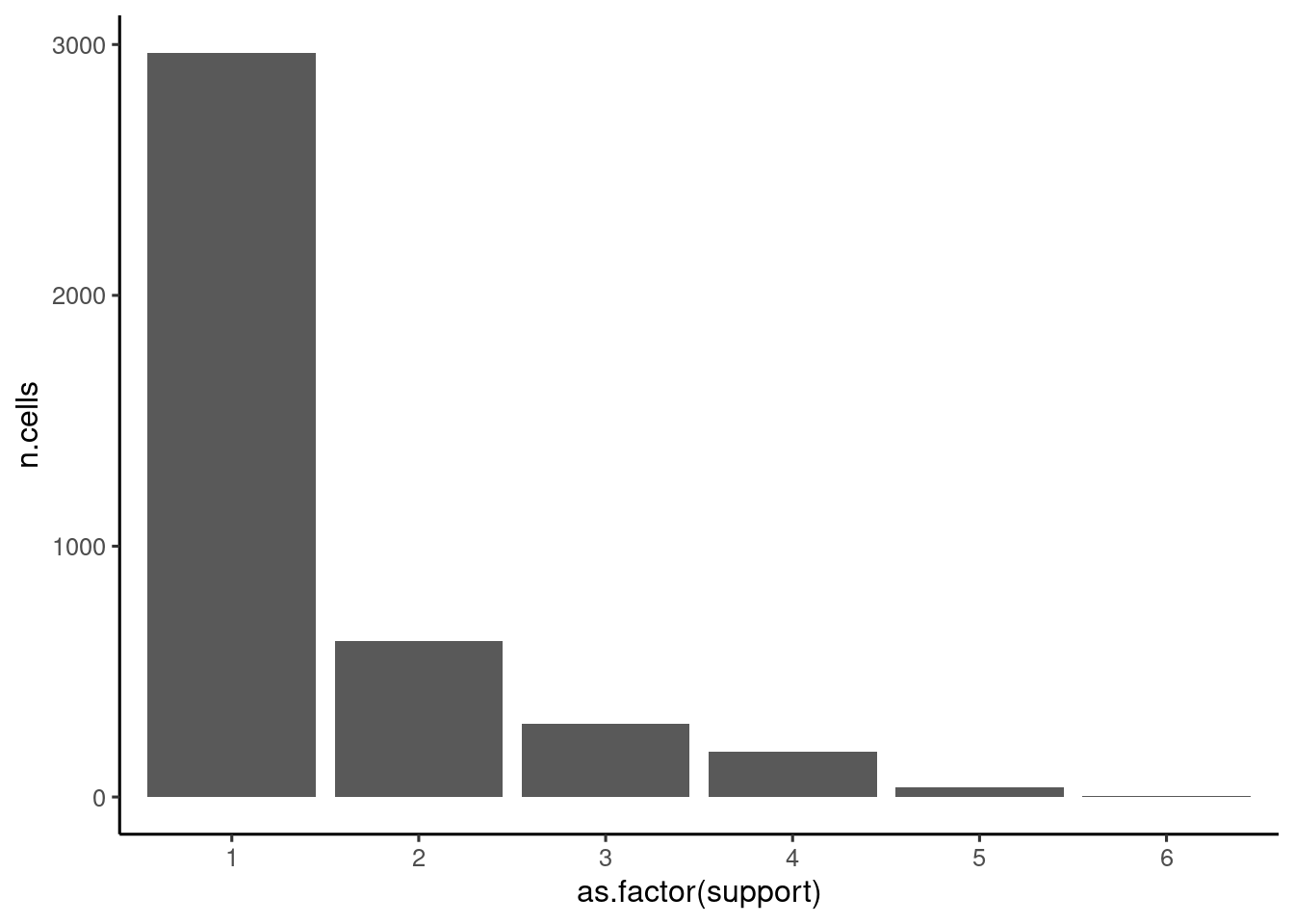
Adjust vireo thresholds
The vireo package uses strict thresholds for assigning cells:
- If prob_max < 0.9 -> “unassigned”
- If prob_doublet_out >= 0.9 -> “doublet”
- If n_vars < 10 -> “unassigned”
However, only 4752 cells had n_vars > 10, if we lower this requirement to n_vars > 5… 9366 cells qualify.
donors <- read.table(paste0(loc, "/hvgs10000/CB-scRNAv31-GEX-lib01_S1_MAF0.01/donor_ids.tsv"),
sep="\t", header=TRUE)
donors$default <- donors$donor_id
# Default except minimum vars = 5 instead of 10
donors$nVars3 <- donors$best_singlet
donors[donors$doublet_logLikRatio >= 0.9, "nVars5"] <- "doublet"
donors[donors$prob_max < 0.9 | donors$n_vars < 3, "nVars3"] <- "unassigned"
# Default except prob_max lowered to 0.7
donors$max0.7 <- donors$best_singlet
donors[donors$doublet_logLikRatio >= 0.9, "max0.7"] <- "doublet"
donors[donors$prob_max < 0.7 | donors$n_vars < 10, "max0.7"] <- "unassigned"
# Default except prob_max lowered to 0.7
donors$allLower <- donors$best_singlet
donors[donors$doublet_logLikRatio >= 0.8, "allLower"] <- "doublet"
donors[donors$prob_max < 0.7 | donors$n_vars < 3, "allLower"] <- "unassigned"
dn <- dplyr::bind_rows(table(donors$default),
table(donors$nVars3),
table(donors$max0.7),
table(donors$allLower))
dn %>% mutate(thresh = c("default", "minVars3", "maxProb0.7", "allLower")) %>%
pivot_longer(-thresh, names_to = "donor", values_to = "n") %>%
filter(donor != "unassigned" & donor != "doublet") %>%
ggplot(aes(x=fct_reorder(thresh, n, .fun = median, .desc=FALSE), y=n, color=donor)) +
geom_jitter(width=0.2, alpha=0.5) +
coord_flip() + xlab("") + theme_classic2(14) +
stat_summary(fun=median, geom="point", shape=18, size=3, color="black")Don't know how to automatically pick scale for object of type table. Defaulting to continuous.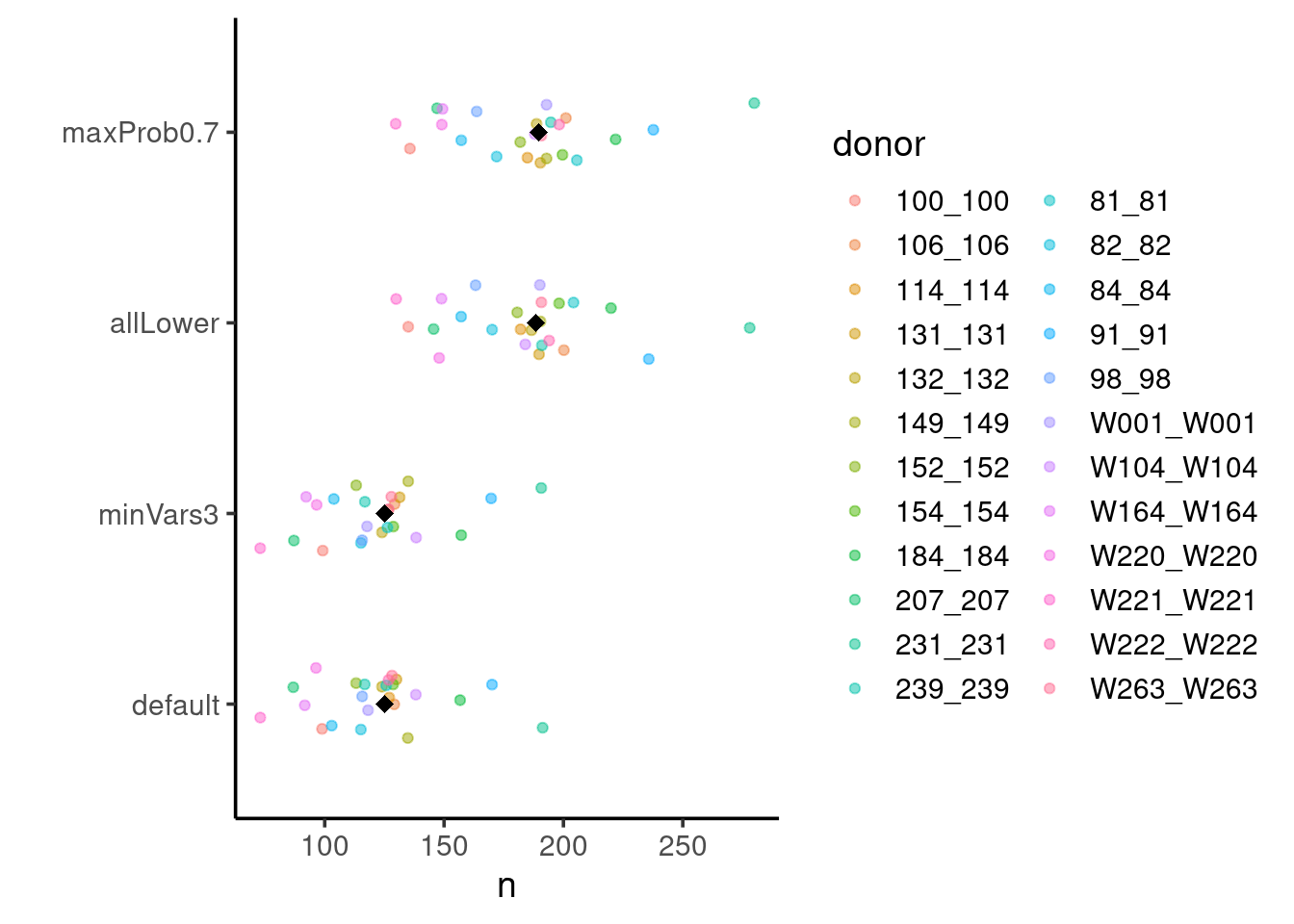
De-novo donor assignment
loc <- "output/pilot2.1_gex/05_vireo"
geno.res.list <- c(paste0(loc, "-noGeno-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.01/"),
paste0(loc, "-noGeno-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.05/"))
id.list <- c("FilterSNPs-HWE0.001-MAF01-Amb100_fixed",
"FilterSNPs-HWE0.001-MAF05-Amb100_fixed")
geno.res <- joinVireoResults(geno.res.list, ids=id.list, d.cols)
geno.res %>%
filter(donor != "unassigned" & donor != "doublet") %>%
ggplot(aes(x=fct_reorder(id, n+1, .fun = median, .desc=FALSE), y=n+1, color=donor)) +
geom_jitter(width=0.2, alpha=0.5) +
coord_flip() + xlab("") +
scale_y_continuous(trans='log2') + theme_classic2(14) +
stat_summary(fun=median, geom="point", shape=18, size=3, color="black") 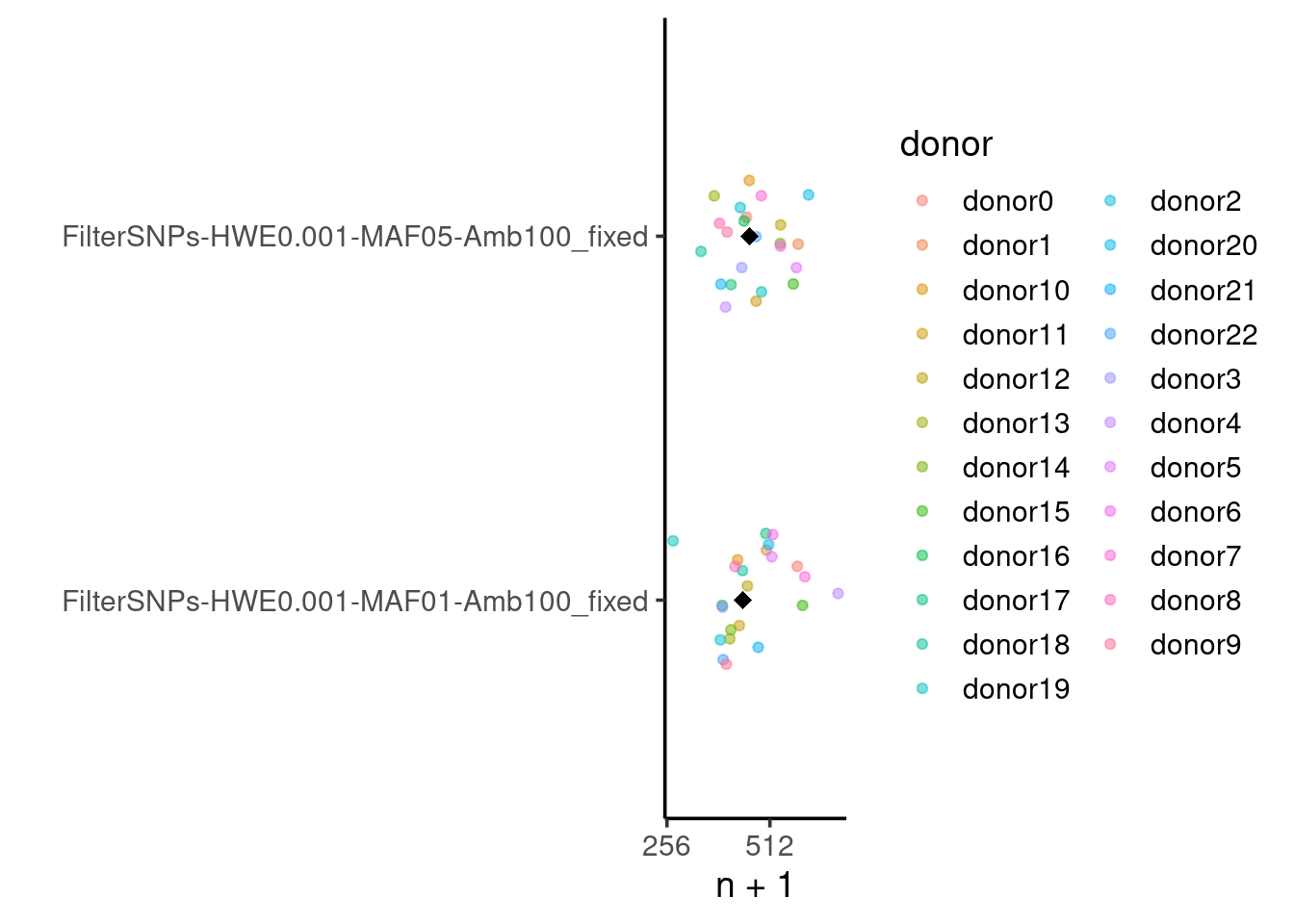
Merging genotype with de novo assignements
Unfortunately, we can not include the common SNPs in a vireo run with donor genotype data provided. However, if it is very clear which de novo donors are which real genotypes, we may be able to use the de novo vireo cell assignments with the real genotypes. To test this we calculated the pairwise correlation in cell assignment probability between de novo and known genotypes (Figure 4).
denovo.assign <- read.table("output/pilot2.1_gex/05_vireo-noGeno-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.01/donor_ids.tsv",
sep="\t", header=TRUE, row.names=1)
denovo.assigned <- row.names(denovo.assign[denovo.assign$donor_id != "unassigned", ])
p.sing.denovo <- read.table("output/pilot2.1_gex/05_vireo-noGeno-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.01/prob_singlet.tsv.gz",
sep="\t", header=TRUE, row.names=1)
p.sing.denovo <- p.sing.denovo[denovo.assigned, ]
p.sing.geno <- read.table("output/pilot2.1_gex/05_vireo/hvgs10000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/prob_singlet.tsv.gz", sep="\t",
header=TRUE, row.names=1)
p.sing.geno <- p.sing.geno[denovo.assigned, ]
p.sing.corr <- cor(p.sing.denovo, p.sing.geno, method="spearman")
corrplot(p.sing.corr, method="color",tl.col="black")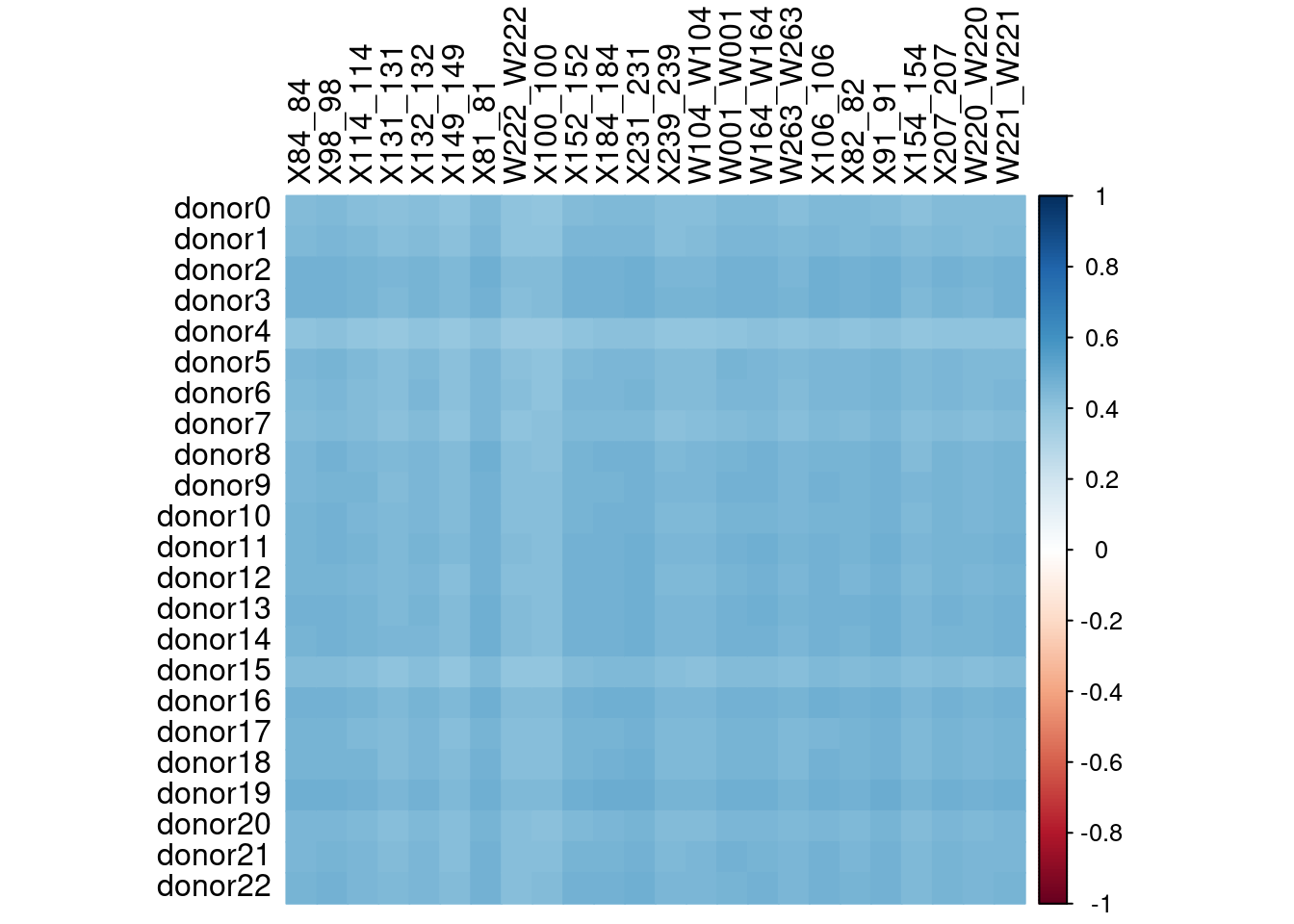
Spearman’s correlation between donor probability scores between from vireo using de novo (rows) and real (columns) genotypes, just including cells that were assigned in the de novo run.
denovo.assign <- read.table("output/pilot2.1_gex/05_vireo-noGeno-SNPfilt/CB-scRNAv31-GEX-lib01_S1_MAF0.01/donor_ids.tsv",
sep="\t", header=TRUE, row.names=1)
geno.assign <- read.table("output/pilot2.1_gex/05_vireo/hvgs10000/CB-scRNAv31-GEX-lib01_S1_MAF0.05/donor_ids.tsv",
sep="\t", header=TRUE, row.names=1)
ovlp.res <- data.frame(geno="x", denovo="x", ovlp=0, ovlp.percent=0)
for(g in unique(geno.assign$donor_id)){
g.cells <- row.names(geno.assign[geno.assign$donor_id == unlist(g), ])
for (d in unique(denovo.assign$donor_id)){
d.cells <-row.names(denovo.assign[denovo.assign$donor_id == unlist(d), ])
ovlp <- length(intersect(g.cells, d.cells))
ovlp.p <- ovlp / length(g.cells)
ovlp.res = rbind(ovlp.res, data.frame(geno=g, denovo=d, ovlp=ovlp, ovlp.percent=ovlp.p))
}
}
ovlp.res.mat <- ovlp.res %>% filter(geno != "x") %>%
pivot_wider(id_cols=geno, names_from = denovo, values_from = ovlp.percent) %>%
column_to_rownames("geno")
corrplot(as.matrix(ovlp.res.mat), method="color",
tl.col="black", col=brewer.pal(n=8, name="YlGnBu"), is.corr=FALSE)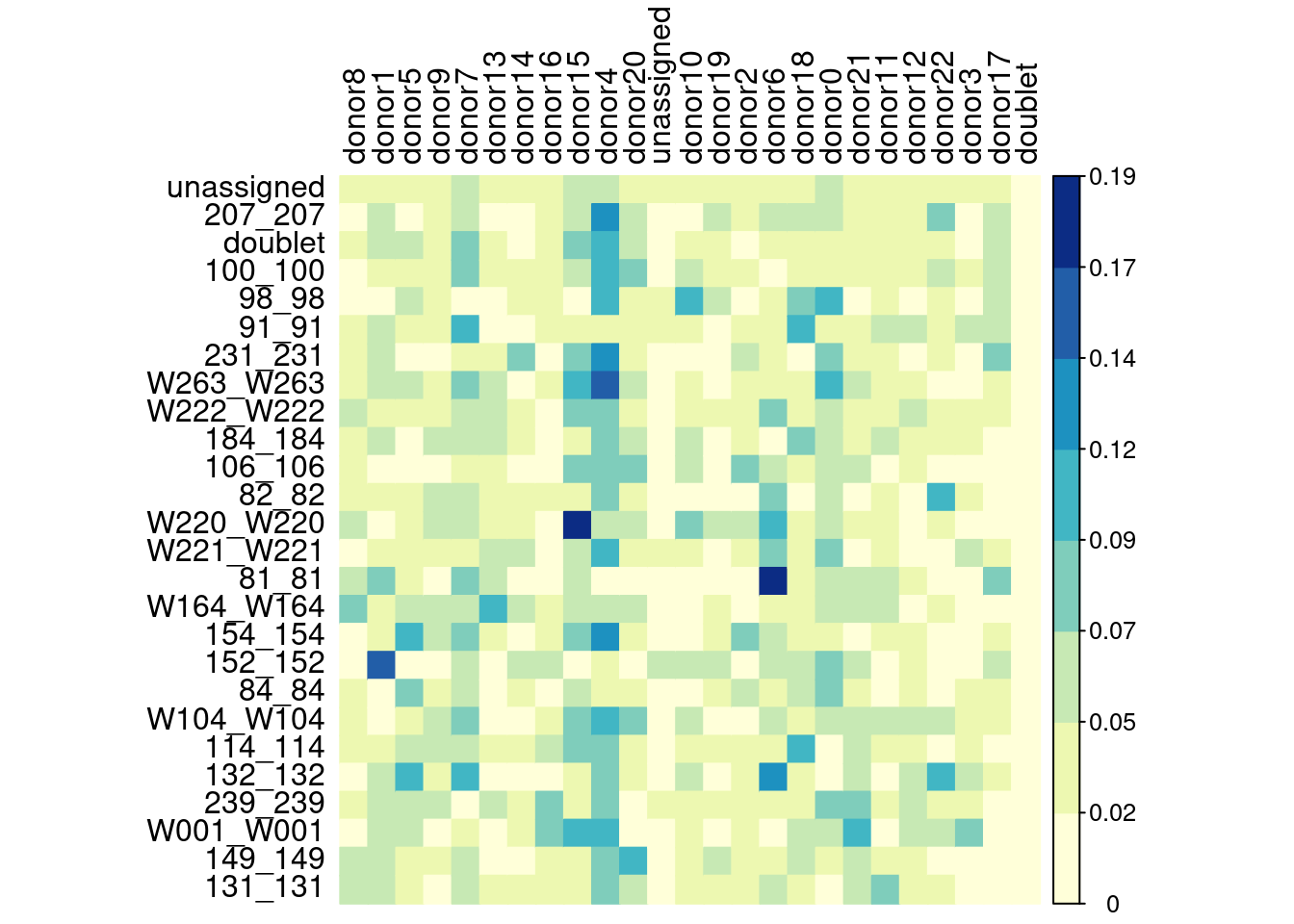
Percent of cells assigned to a genotype that were assigned to a de-novo genotype. Where a de-novo genotype would be considered a match with a real genotype if most cells from a donor were assigned to a single de-novo genotype.
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.0.4 (2021-02-15)
os Red Hat Enterprise Linux
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2021-08-06
─ Packages ───────────────────────────────────────────────────────────────────
package * version date lib source
abind 1.4-5 2016-07-21 [1] CRAN (R 4.0.2)
assertthat 0.2.1 2019-03-21 [1] CRAN (R 4.0.2)
backports 1.2.1 2020-12-09 [1] CRAN (R 4.0.4)
broom 0.7.8 2021-06-24 [1] CRAN (R 4.0.4)
bslib 0.2.5.1 2021-05-18 [1] CRAN (R 4.0.4)
cachem 1.0.5 2021-05-15 [1] CRAN (R 4.0.4)
callr 3.7.0 2021-04-20 [1] CRAN (R 4.0.4)
car 3.0-11 2021-06-27 [1] CRAN (R 4.0.4)
carData 3.0-4 2020-05-22 [1] CRAN (R 4.0.2)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.0.2)
cli 3.0.0 2021-06-30 [1] CRAN (R 4.0.4)
colorspace 2.0-2 2021-06-24 [1] CRAN (R 4.0.4)
corrplot * 0.90 2021-06-30 [1] CRAN (R 4.0.4)
crayon 1.4.1 2021-02-08 [1] CRAN (R 4.0.4)
curl 4.3.2 2021-06-23 [1] CRAN (R 4.0.4)
data.table 1.14.0 2021-02-21 [1] CRAN (R 4.0.4)
DBI 1.1.1 2021-01-15 [1] CRAN (R 4.0.4)
dbplyr 2.1.1 2021-04-06 [1] CRAN (R 4.0.4)
desc 1.3.0 2021-03-05 [1] CRAN (R 4.0.4)
devtools 2.4.2 2021-06-07 [1] CRAN (R 4.0.4)
digest 0.6.27 2020-10-24 [1] CRAN (R 4.0.2)
dplyr * 1.0.7 2021-06-18 [1] CRAN (R 4.0.4)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.0.4)
evaluate 0.14 2019-05-28 [1] CRAN (R 4.0.2)
fansi 0.5.0 2021-05-25 [1] CRAN (R 4.0.4)
farver 2.1.0 2021-02-28 [1] CRAN (R 4.0.4)
fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.0.3)
forcats * 0.5.1 2021-01-27 [1] CRAN (R 4.0.4)
foreign 0.8-81 2020-12-22 [2] CRAN (R 4.0.4)
fs 1.5.0 2020-07-31 [1] CRAN (R 4.0.2)
generics 0.1.0 2020-10-31 [1] CRAN (R 4.0.2)
ggplot2 * 3.3.5 2021-06-25 [1] CRAN (R 4.0.4)
ggpubr * 0.4.0 2020-06-27 [1] CRAN (R 4.0.3)
ggsignif 0.6.2 2021-06-14 [1] CRAN (R 4.0.4)
ggupset * 0.3.0 2020-05-05 [1] CRAN (R 4.0.4)
git2r 0.28.0 2021-01-10 [1] CRAN (R 4.0.4)
glue 1.4.2 2020-08-27 [1] CRAN (R 4.0.2)
gtable 0.3.0 2019-03-25 [1] CRAN (R 4.0.2)
haven 2.4.1 2021-04-23 [1] CRAN (R 4.0.4)
highr 0.9 2021-04-16 [1] CRAN (R 4.0.4)
hms 1.1.0 2021-05-17 [1] CRAN (R 4.0.4)
htmltools 0.5.1.1 2021-01-22 [1] CRAN (R 4.0.3)
httpuv 1.6.1 2021-05-07 [1] CRAN (R 4.0.4)
httr 1.4.2 2020-07-20 [1] CRAN (R 4.0.2)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.0.4)
jsonlite 1.7.2 2020-12-09 [1] CRAN (R 4.0.4)
knitr 1.33 2021-04-24 [1] CRAN (R 4.0.4)
labeling 0.4.2 2020-10-20 [1] CRAN (R 4.0.2)
later 1.2.0 2021-04-23 [1] CRAN (R 4.0.4)
lifecycle 1.0.0 2021-02-15 [1] CRAN (R 4.0.4)
lubridate 1.7.10 2021-02-26 [1] CRAN (R 4.0.4)
magrittr 2.0.1 2020-11-17 [1] CRAN (R 4.0.3)
memoise 2.0.0 2021-01-26 [1] CRAN (R 4.0.4)
modelr 0.1.8 2020-05-19 [1] CRAN (R 4.0.2)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.0.2)
openxlsx 4.2.4 2021-06-16 [1] CRAN (R 4.0.4)
pillar 1.6.1 2021-05-16 [1] CRAN (R 4.0.4)
pkgbuild 1.2.0 2020-12-15 [1] CRAN (R 4.0.4)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.0.2)
pkgload 1.2.1 2021-04-06 [1] CRAN (R 4.0.4)
prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.0.2)
processx 3.5.2 2021-04-30 [1] CRAN (R 4.0.4)
promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.0.4)
ps 1.6.0 2021-02-28 [1] CRAN (R 4.0.4)
purrr * 0.3.4 2020-04-17 [1] CRAN (R 4.0.2)
R6 2.5.0 2020-10-28 [1] CRAN (R 4.0.2)
RColorBrewer * 1.1-2 2014-12-07 [1] CRAN (R 4.0.2)
Rcpp 1.0.7 2021-07-07 [1] CRAN (R 4.0.4)
readr * 1.4.0 2020-10-05 [1] CRAN (R 4.0.2)
readxl 1.3.1 2019-03-13 [1] CRAN (R 4.0.2)
remotes 2.4.0 2021-06-02 [1] CRAN (R 4.0.4)
reprex 2.0.0 2021-04-02 [1] CRAN (R 4.0.4)
rio 0.5.27 2021-06-21 [1] CRAN (R 4.0.4)
rlang 0.4.11 2021-04-30 [1] CRAN (R 4.0.4)
rmarkdown 2.9 2021-06-15 [1] CRAN (R 4.0.4)
rprojroot 2.0.2 2020-11-15 [1] CRAN (R 4.0.3)
rstatix 0.7.0 2021-02-13 [1] CRAN (R 4.0.4)
rstudioapi 0.13 2020-11-12 [1] CRAN (R 4.0.3)
rvest 1.0.0 2021-03-09 [1] CRAN (R 4.0.4)
sass 0.4.0 2021-05-12 [1] CRAN (R 4.0.4)
scales 1.1.1 2020-05-11 [1] CRAN (R 4.0.2)
sessioninfo 1.1.1 2018-11-05 [1] CRAN (R 4.0.2)
stringi 1.7.2 2021-07-14 [1] CRAN (R 4.0.4)
stringr * 1.4.0 2019-02-10 [1] CRAN (R 4.0.2)
testthat 3.0.4 2021-07-01 [1] CRAN (R 4.0.4)
tibble * 3.1.2 2021-05-16 [1] CRAN (R 4.0.4)
tidyr * 1.1.3 2021-03-03 [1] CRAN (R 4.0.4)
tidyselect 1.1.1 2021-04-30 [1] CRAN (R 4.0.4)
tidyverse * 1.3.1 2021-04-15 [1] CRAN (R 4.0.4)
usethis 2.0.1 2021-02-10 [1] CRAN (R 4.0.4)
utf8 1.2.1 2021-03-12 [1] CRAN (R 4.0.4)
vctrs 0.3.8 2021-04-29 [1] CRAN (R 4.0.4)
withr 2.4.2 2021-04-18 [1] CRAN (R 4.0.4)
workflowr 1.6.2 2020-04-30 [1] CRAN (R 4.0.2)
xfun 0.24 2021-06-15 [1] CRAN (R 4.0.4)
xml2 1.3.2 2020-04-23 [1] CRAN (R 4.0.2)
yaml 2.2.1 2020-02-01 [1] CRAN (R 4.0.2)
zip 2.2.0 2021-05-31 [1] CRAN (R 4.0.4)
[1] /mnt/mcfiles/cazodi/R/x86_64-pc-linux-gnu-library/4.0
[2] /opt/R/4.0.4/lib/R/library