BAUH: MND scRNA-seq pilot 2: DropletQC
C.B. Azodi
2021-09-28
Last updated: 2021-09-28
Checks: 5 2
Knit directory: BAUH_2020_MND-single-cell/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/possorted_genome_bam.bam | output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/possorted_genome_bam.bam |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/raw_non_zeroUMI_barcodes.tsv | output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/raw_non_zeroUMI_barcodes.tsv |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version c703e21. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Untracked files:
Untracked: .snakemake/
Untracked: 2021-04-27_pilot2_nCells-per-donor.pdf
Untracked: 2021-08-03_pilot2_nCells-per-donor.pdf
Untracked: BAUH_2020_MND-single-cell.Rproj
Untracked: GRCh38_turboGFP-RFP_reference/
Untracked: Log.out
Untracked: Rplots.pdf
Untracked: analysis/2021-09-28_pilot2_CellCalling-comparison.Rmd
Untracked: star-help.txt
Untracked: test/
Untracked: test_learn/
Untracked: test_maf01_notFiltered/
Untracked: test_maf05/
Untracked: test_noGeno/
Untracked: test_vireo/
Untracked: workflow/dropkick_get_ambient_and_hvgs.py
Unstaged changes:
Modified: analysis/2021-08-04_DropletQC.Rmd
Modified: analysis/index.Rmd
Modified: workflow/config_ambient_pilot2.1.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/2021-08-04_DropletQC.Rmd) and HTML (public/2021-08-04_DropletQC.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | c703e21 | cazodi | 2021-09-27 | added workflowr pages for cell calling with emptydroplet and dropkick |
| html | c703e21 | cazodi | 2021-09-27 | added workflowr pages for cell calling with emptydroplet and dropkick |
| Rmd | 679ded7 | cazodi | 2021-08-06 | updates for davis |
| html | 679ded7 | cazodi | 2021-08-06 | updates for davis |
#devtools::install_github("powellgenomicslab/DropletQC", build_vignettes = TRUE)
suppressPackageStartupMessages({
library(DropletQC)
library(DropletUtils)
library(ggplot2)
library(ggpubr)
library(patchwork)
})
rerun <- FALSE
out.dir <- "output/pilot2.1_gex/06_dropletQC/"DropletQC
A new droplet single-cell QC package from Joseph Powell’s lab that is able to detect empty droplets, damaged, and intact cells, and accurately distinguish from one another. This approach is based on a novel quality control metric, the nuclear fraction, which quantifies for each droplet the fraction of RNA originating from unspliced, nuclear pre-mRNA ( preprint).
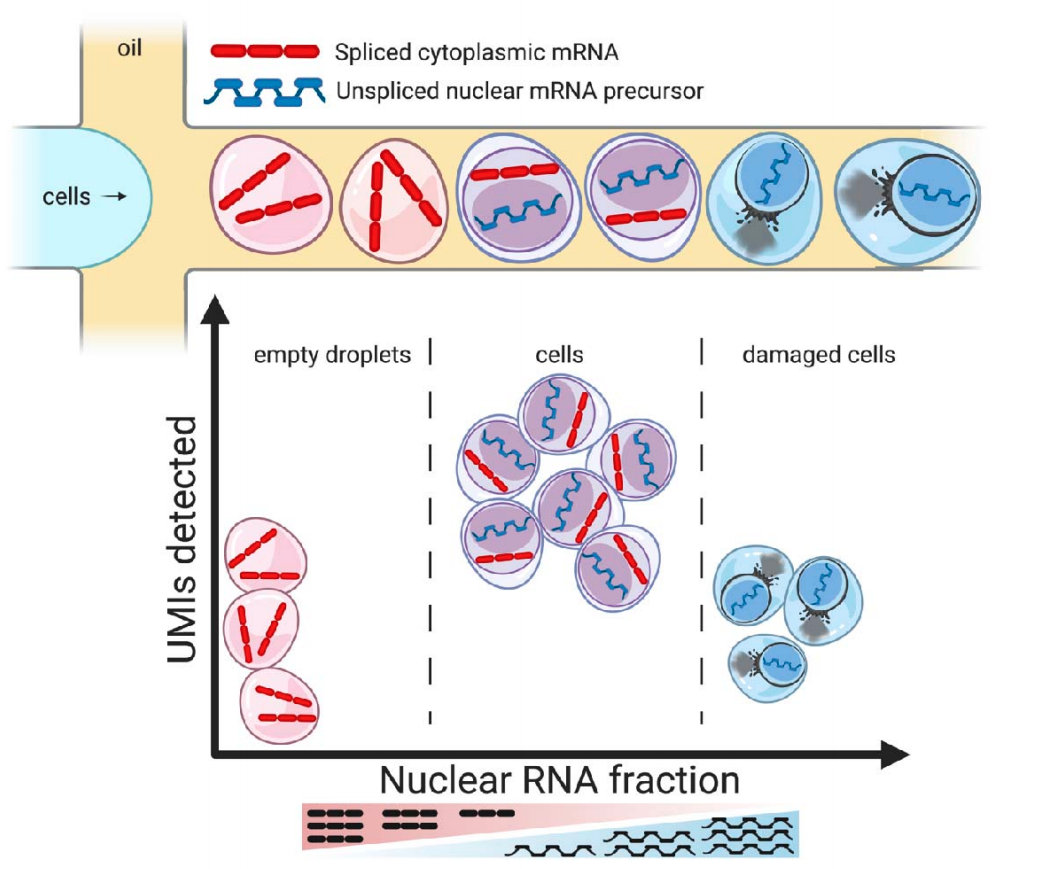
DropletQC theory
cellranger called cells
Because I processed the data with CellRanger counts, the BAM file contains region tags which identify aligned reads as intronic or exonic. That means we can calculate the nuclear fraction directly from the bam file:
nuclear fraction = intronic reads / (intronic reads + exonic reads)
To identify empty and then damaged barcodes, I need to generate an input that contains the nuclear fraction in the first column and the total UMI count in the second column. Their function to calcuate nuclear_fraction_tags, only produces the nf, so I have to do this manually. I removed barcodes that had a UMI count of zero, leaving 1,544,656 barcodes.
if(rerun) {
# Note this takes about 150GB memory and 1 hour
out.dir <- "output/pilot2.1_gex/06_dropletQC/"
in.dir <- "output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs"
umi <- read10xCounts(paste0(in.dir, "/raw_feature_bc_matrix/"))
umi <- data.frame(id = colData(umi)$Barcode, umi = colSums(counts(umi)))
row.names(umi) <- umi$id
umi <- subset(umi, umi > 0)
write.table(umi$id, "output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/raw_non_zeroUMI_barcodes.tsv", row.names = FALSE, col.names = FALSE, quote=FALSE)
gff.file <- "/mnt/beegfs/mccarthy/scratch/general/cazodi/Datasets/references/human/hg38.98_turboGFP/Homo_sapiens.GRCh38.93.filtered_turboGFP.gtf"
bam.file <- "/mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/possorted_genome_bam.bam"
bc.file <- "/mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2.1_gex/01_cellranger/CB-scRNAv31-GEX-lib01_S1/outs/raw_non_zeroUMI_barcodes.tsv"
nf <- nuclear_fraction_annotation(annotation_path = gff.file,
bam = bam.file, barcodes = bc.file,
tiles = 1, cores = 1, verbose = FALSE)
sce.stats <- merge(nf, umi, by="row.names")
row.names(sce.stats) <- sce.stats$id
sce.stats[, c("Row.names", "id")] <- NULL
saveRDS(sce.stats, paste0(out.dir, "lib01_cellrangerRAW_nf-umi.rds"))
} else{
sce.stats <- readRDS(paste0(out.dir, "lib01_cellrangerRAW_nf-umi.rds"))
}
head(sce.stats) nuclear_fraction umi
AAACCCAAGAAACCAT-1 0.2727273 1
AAACCCAAGAAACCCG-1 0.0000000 1
AAACCCAAGAAACTAC-1 0.6666667 1
AAACCCAAGAAAGTCT-1 0.5000000 1
AAACCCAAGAAATCCA-1 0.2727273 2
AAACCCAAGAACAAGG-1 0.0000000 1Once the nuclear fraction score has been calculated for barcodes with at least one UMI, the identify_empty_drops and identify_damaged_cells functions were applied to identify each these populations (note that empty or damaged cells are flagged, not removed!).
sce.stats <- identify_empty_drops(sce.stats, include_plot = TRUE)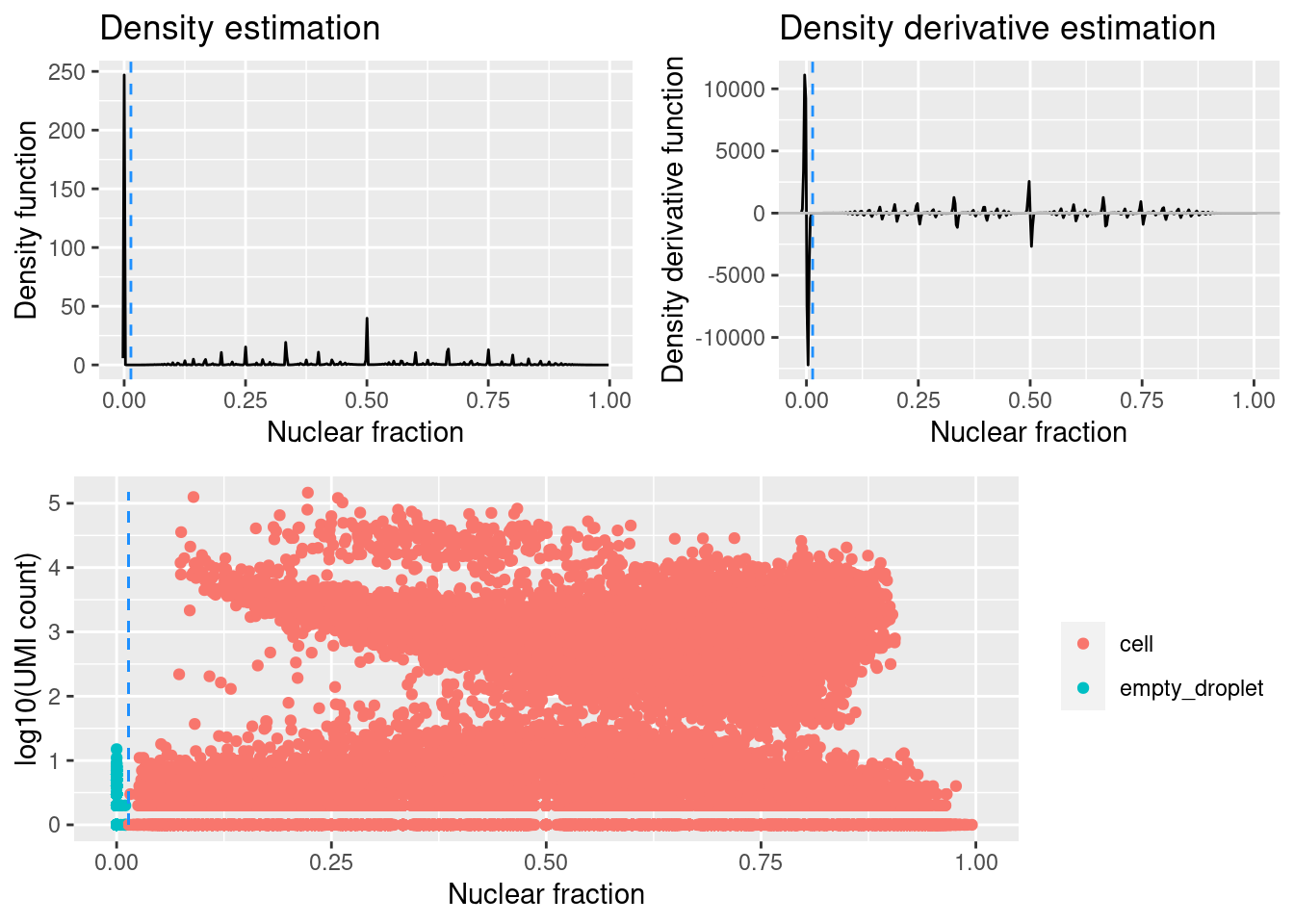
Plots output by identify_empty_droplets. The dashed blue lines mark where the two rescue parameters are set (i.e. at which barcodes are called as cells).
table(sce.stats$cell_status)
cell empty_droplet
898351 646305 Next we can identify damaged cells. Intuitively, empty droplets have a low RNA content and low nuclear fraction score (bottom left). Damaged cells have a low RNA content and high nuclear fraction score (bottom right). However, running DropletQC on our raw data, no damaged cells are called, even though many are located in that bottom right quadrant of the plot. According to the package vignette, performance is improved if cell type information is provided. Because we hope all of our barcodes are either empty or motor neurons, this might not make a difference, but hard to tell. Think about trying to run DropletQC on data after doing an initial, loose filtering for cells and after donor ID and cell annotation. Maybe it will work better at that stage!
sce.stats$celltype <- "mn"
sce.stats.dc <- identify_damaged_cells(sce.stats, output_plots=TRUE)[1] "The following cell types were provided; mn"
[1] "Fitting models with EM"
fitting ...
|
| | 0%
|
|======================= | 33%
|
|=============================================== | 67%
|
|======================================================================| 100%
[1] "Creating requested plots"table(sce.stats.dc[[1]]$cell_status)
cell empty_droplet
898351 646305 ggplot(sce.stats.dc[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_classic2()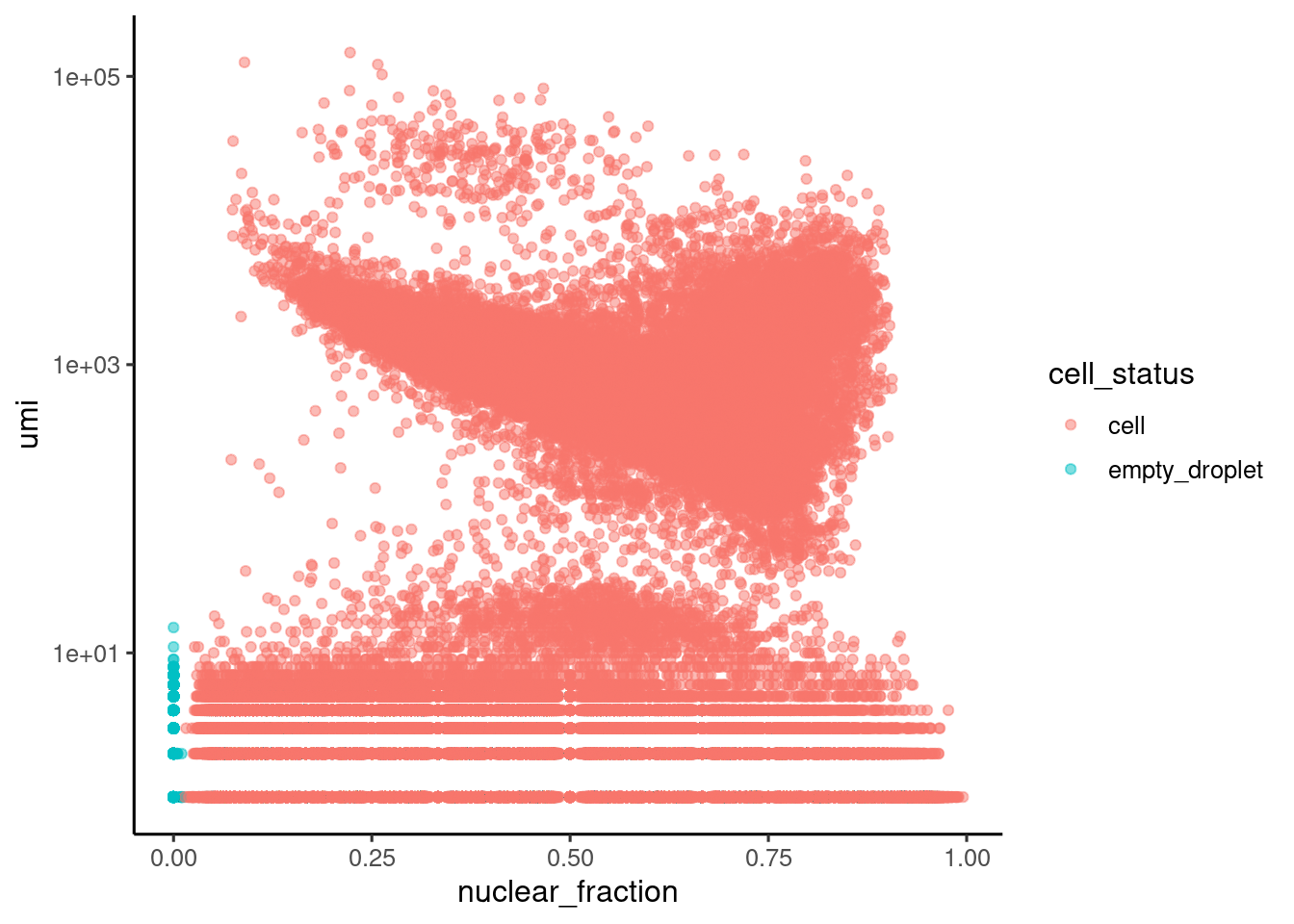
| Version | Author | Date |
|---|---|---|
| 679ded7 | cazodi | 2021-08-06 |
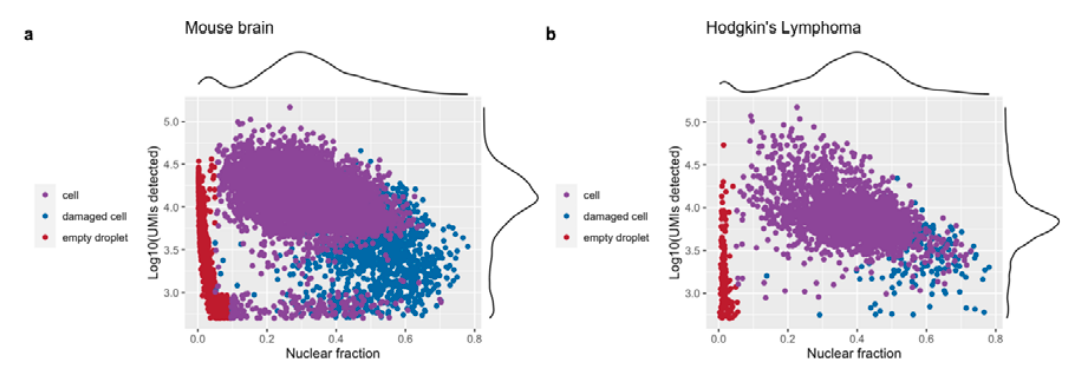
Examples from DropletQC paper
For each provided cell type the left plot marks barcodes called as cells or damaged cells - excluding any empty droplets. The remaining plots illustrate the Guassian distribution/s fit to the nculear fraction (centre) and log10(UMI count) (right) using expectation maximisation. Similar to the identify_empty_drops function, the identify_damaged_cells function inlcudes two rescue parameters; nf_sep and umi_sep_perc. For a population of barcodes to be called as damaged cells:
The mean of the distribution fit to the nuclear fraction (vertical solid red line) must be at least
nf_sep(default 0.15) greater than the mean of the cell population - the threshold marked by the dashed blue lineThe mean of the distribution fit to the log10(UMI counts) (vertical solid red line) must be at least
umi_sep_perc(default 50%) percent less than the mean of the cell population - threshold indicated by the dashed blue line
The ability to detect damaged cells will depend on the the accuracy of the cell type annotation. Different cell types or states can contain varying amounts of nuclear or total RNA, and may cause mixed populations of cells to be mislabeled as damaged.
wrap_plots(sce.stats.dc[[2]], nrow = 1)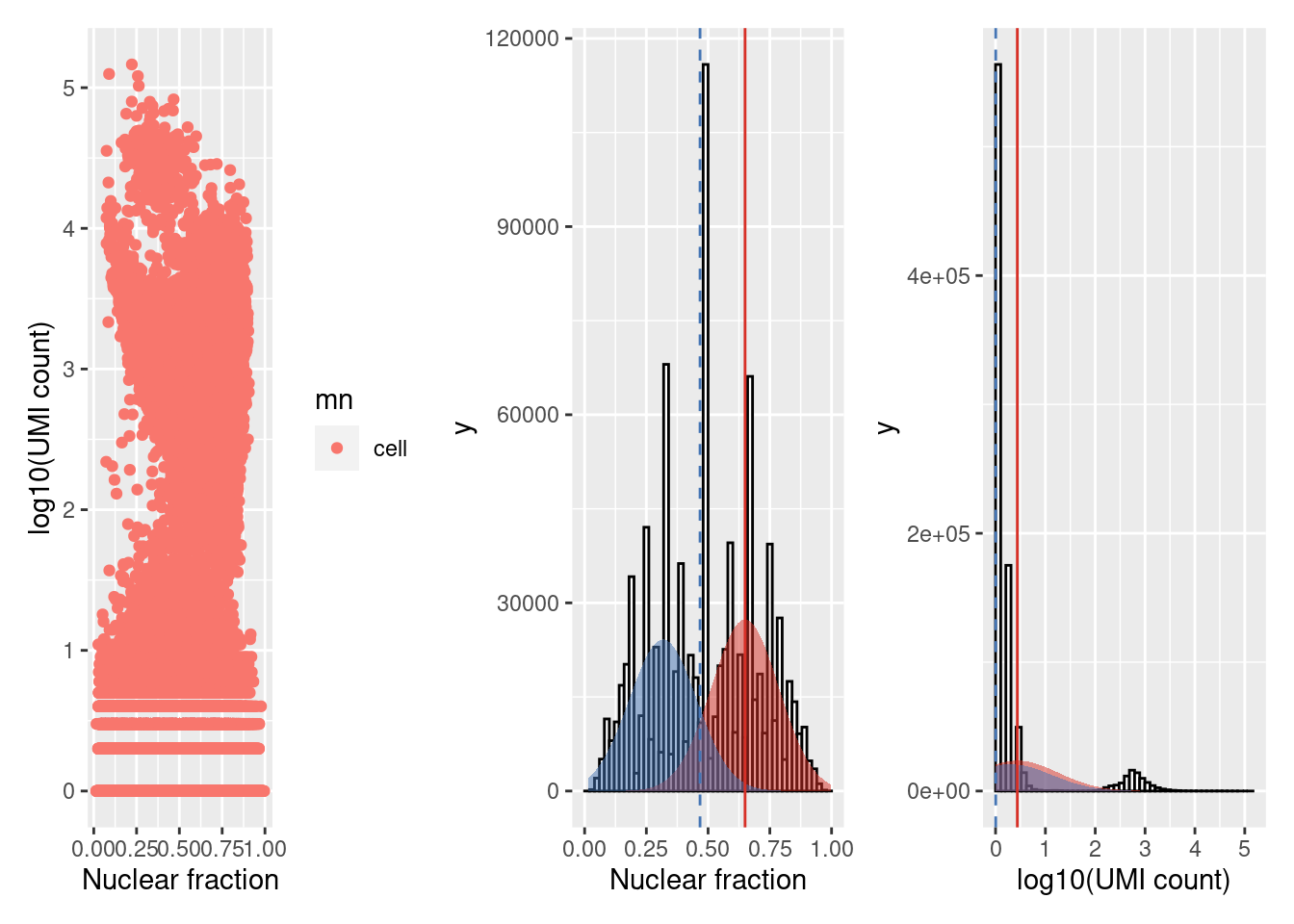
| Version | Author | Date |
|---|---|---|
| 679ded7 | cazodi | 2021-08-06 |
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.0.4 (2021-02-15)
os Rocky Linux 8.4 (Green Obsidian)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2021-09-28
─ Packages ───────────────────────────────────────────────────────────────────
package * version date lib
abind 1.4-5 2016-07-21 [1]
assertthat 0.2.1 2019-03-21 [1]
backports 1.2.1 2020-12-09 [1]
beachmat 2.6.4 2020-12-20 [1]
Biobase * 2.50.0 2020-10-27 [1]
BiocGenerics * 0.36.1 2021-04-16 [1]
BiocParallel 1.24.1 2020-11-06 [1]
bitops 1.0-7 2021-04-24 [1]
broom 0.7.9 2021-07-27 [1]
bslib 0.2.5.1 2021-05-18 [1]
cachem 1.0.6 2021-08-19 [1]
callr 3.7.0 2021-04-20 [1]
car 3.0-11 2021-06-27 [1]
carData 3.0-4 2020-05-22 [1]
cellranger 1.1.0 2016-07-27 [1]
cli 3.0.1 2021-07-17 [1]
colorspace 2.0-2 2021-06-24 [1]
cowplot 1.1.1 2020-12-30 [1]
crayon 1.4.1 2021-02-08 [1]
curl 4.3.2 2021-06-23 [1]
data.table 1.14.2 2021-09-27 [1]
DBI 1.1.1 2021-01-15 [1]
DelayedArray 0.16.3 2021-03-24 [1]
DelayedMatrixStats 1.12.3 2021-02-03 [1]
desc 1.3.0 2021-03-05 [1]
devtools 2.4.2 2021-06-07 [1]
digest 0.6.28 2021-09-23 [1]
dplyr 1.0.7 2021-06-18 [1]
dqrng 0.3.0 2021-05-01 [1]
DropletQC * 0.0.0.9000 2021-09-27 [1]
DropletUtils * 1.10.3 2021-02-02 [1]
edgeR 3.32.1 2021-01-14 [1]
ellipsis 0.3.2 2021-04-29 [1]
evaluate 0.14 2019-05-28 [1]
fansi 0.5.0 2021-05-25 [1]
farver 2.1.0 2021-02-28 [1]
fastmap 1.1.0 2021-01-25 [1]
forcats 0.5.1 2021-01-27 [1]
foreign 0.8-81 2020-12-22 [2]
fs 1.5.0 2020-07-31 [1]
generics 0.1.0 2020-10-31 [1]
GenomeInfoDb * 1.26.7 2021-04-08 [1]
GenomeInfoDbData 1.2.4 2020-11-10 [1]
GenomicRanges * 1.42.0 2020-10-27 [1]
ggplot2 * 3.3.5 2021-06-25 [1]
ggpubr * 0.4.0 2020-06-27 [1]
ggsignif 0.6.2 2021-06-14 [1]
git2r 0.28.0 2021-01-10 [1]
glue 1.4.2 2020-08-27 [1]
gtable 0.3.0 2019-03-25 [1]
haven 2.4.3 2021-08-04 [1]
HDF5Array 1.18.1 2021-02-04 [1]
highr 0.9 2021-04-16 [1]
hms 1.1.0 2021-05-17 [1]
htmltools 0.5.2 2021-08-25 [1]
httpuv 1.6.2 2021-08-18 [1]
IRanges * 2.24.1 2020-12-12 [1]
jquerylib 0.1.4 2021-04-26 [1]
jsonlite 1.7.2 2020-12-09 [1]
KernSmooth 2.23-20 2021-05-03 [1]
knitr 1.34 2021-09-09 [1]
ks 1.13.2 2021-07-01 [1]
labeling 0.4.2 2020-10-20 [1]
later 1.3.0 2021-08-18 [1]
lattice 0.20-41 2020-04-02 [2]
lifecycle 1.0.1 2021-09-24 [1]
limma 3.46.0 2020-10-27 [1]
locfit 1.5-9.4 2020-03-25 [1]
magrittr 2.0.1 2020-11-17 [1]
Matrix 1.3-4 2021-06-01 [1]
MatrixGenerics * 1.2.1 2021-01-30 [1]
matrixStats * 0.60.0 2021-07-26 [1]
mclust 5.4.7 2020-11-20 [1]
memoise 2.0.0 2021-01-26 [1]
munsell 0.5.0 2018-06-12 [1]
mvtnorm 1.1-2 2021-06-07 [1]
openxlsx 4.2.4 2021-06-16 [1]
patchwork * 1.1.1 2020-12-17 [1]
pillar 1.6.3 2021-09-26 [1]
pkgbuild 1.2.0 2020-12-15 [1]
pkgconfig 2.0.3 2019-09-22 [1]
pkgload 1.2.2 2021-09-11 [1]
pracma 2.3.3 2021-01-23 [1]
prettyunits 1.1.1 2020-01-24 [1]
processx 3.5.2 2021-04-30 [1]
promises 1.2.0.1 2021-02-11 [1]
ps 1.6.0 2021-02-28 [1]
purrr 0.3.4 2020-04-17 [1]
R.methodsS3 1.8.1 2020-08-26 [1]
R.oo 1.24.0 2020-08-26 [1]
R.utils 2.10.1 2020-08-26 [1]
R6 2.5.1 2021-08-19 [1]
Rcpp 1.0.7 2021-07-07 [1]
RCurl 1.98-1.4 2021-08-17 [1]
readxl 1.3.1 2019-03-13 [1]
remotes 2.4.0 2021-06-02 [1]
rhdf5 2.34.0 2020-10-27 [1]
rhdf5filters 1.2.1 2021-05-03 [1]
Rhdf5lib 1.12.1 2021-01-26 [1]
rio 0.5.27 2021-06-21 [1]
rlang 0.4.11 2021-04-30 [1]
rmarkdown 2.11 2021-09-14 [1]
rprojroot 2.0.2 2020-11-15 [1]
rstatix 0.7.0 2021-02-13 [1]
rstudioapi 0.13 2020-11-12 [1]
S4Vectors * 0.28.1 2020-12-09 [1]
sass 0.4.0 2021-05-12 [1]
scales 1.1.1 2020-05-11 [1]
scuttle 1.0.4 2020-12-17 [1]
sessioninfo 1.1.1 2018-11-05 [1]
SingleCellExperiment * 1.12.0 2020-10-27 [1]
sparseMatrixStats 1.2.1 2021-02-02 [1]
stringi 1.7.4 2021-08-25 [1]
stringr 1.4.0 2019-02-10 [1]
SummarizedExperiment * 1.20.0 2020-10-27 [1]
testthat 3.0.4 2021-07-01 [1]
tibble 3.1.4 2021-08-25 [1]
tidyr 1.1.4 2021-09-27 [1]
tidyselect 1.1.1 2021-04-30 [1]
usethis 2.0.1 2021-02-10 [1]
utf8 1.2.2 2021-07-24 [1]
vctrs 0.3.8 2021-04-29 [1]
whisker 0.4 2019-08-28 [1]
withr 2.4.2 2021-04-18 [1]
workflowr 1.6.2 2020-04-30 [1]
xfun 0.26 2021-09-14 [1]
XVector 0.30.0 2020-10-27 [1]
yaml 2.2.1 2020-02-01 [1]
zip 2.2.0 2021-05-31 [1]
zlibbioc 1.36.0 2020-10-27 [1]
source
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Github (powellgenomicslab/DropletQC@4409c95)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.2)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
[1] /mnt/mcfiles/cazodi/R/x86_64-pc-linux-gnu-library/4.0
[2] /opt/R/4.0.4/lib/R/library