BAUH: MND scRNA-seq pilot 2: EmptyDroplet
Christina B. Azodi
2021-09-27
Last updated: 2021-09-27
Checks: 6 1
Knit directory: BAUH_2020_MND-single-cell/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e5d517e. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Untracked files:
Untracked: .snakemake/
Untracked: 2021-04-27_pilot2_nCells-per-donor.pdf
Untracked: 2021-08-03_pilot2_nCells-per-donor.pdf
Untracked: BAUH_2020_MND-single-cell.Rproj
Untracked: GRCh38_turboGFP-RFP_reference/
Untracked: Log.out
Untracked: Rplots.pdf
Untracked: analysis/2021-08-03_BAUH_MND-pilot-2.Rmd
Untracked: analysis/2021-09-27_pilot2_EmptyDrops.Rmd
Untracked: star-help.txt
Untracked: test/
Untracked: test_learn/
Untracked: test_maf01_notFiltered/
Untracked: test_maf05/
Untracked: test_noGeno/
Untracked: test_vireo/
Untracked: workflow/dropkick_get_ambient_and_hvgs.py
Unstaged changes:
Modified: analysis/2021-07-07_geno-guided-donor-assignment-tests.Rmd
Modified: analysis/2021-07-07_geno-guided-donor-assignment-tests.html
Modified: analysis/index.Rmd
Modified: workflow/config_ambient_pilot2.1.yml
Staged changes:
Modified: analysis/2021-07-07_geno-guided-donor-assignment-tests.Rmd
New: analysis/2021-07-07_geno-guided-donor-assignment-tests.html
Modified: analysis/2021-08-04_DropletQC.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
suppressPackageStartupMessages({
library(tidyverse)
library(knitr)
library(Matrix)
library(jcolors)
library(DropletUtils)
library(viridis)
})Overview
Cell calling
CellRanger & EmptyDroplet
CellRanger count was used to assign reads to cells based on the cell barcode, aligns raw reads to the genome, quantifies counts, and corrects for PCR amplification bias by converting to UMI counts per gene per barcode. CellRanger also applies EmptyDroplet (Lun et al., 2018) to call barcodes as cell or background. It does this by identifying significant deviations from the expression profile of the ambient solution, with a hard lower UMI threshold set to 100. In pilot #1, the UMI counts per cell quickly drops (i.e. a sharp knee) to the hundreds, making a more clear cut of point for cell calling by EmptyDroplet (except for S3, which we discussed is likely due to poor cell quality). This steep drop-off indicates one population of healthy, good quality cells with many reads (left) and one population of empty droplets with very few reads (right).
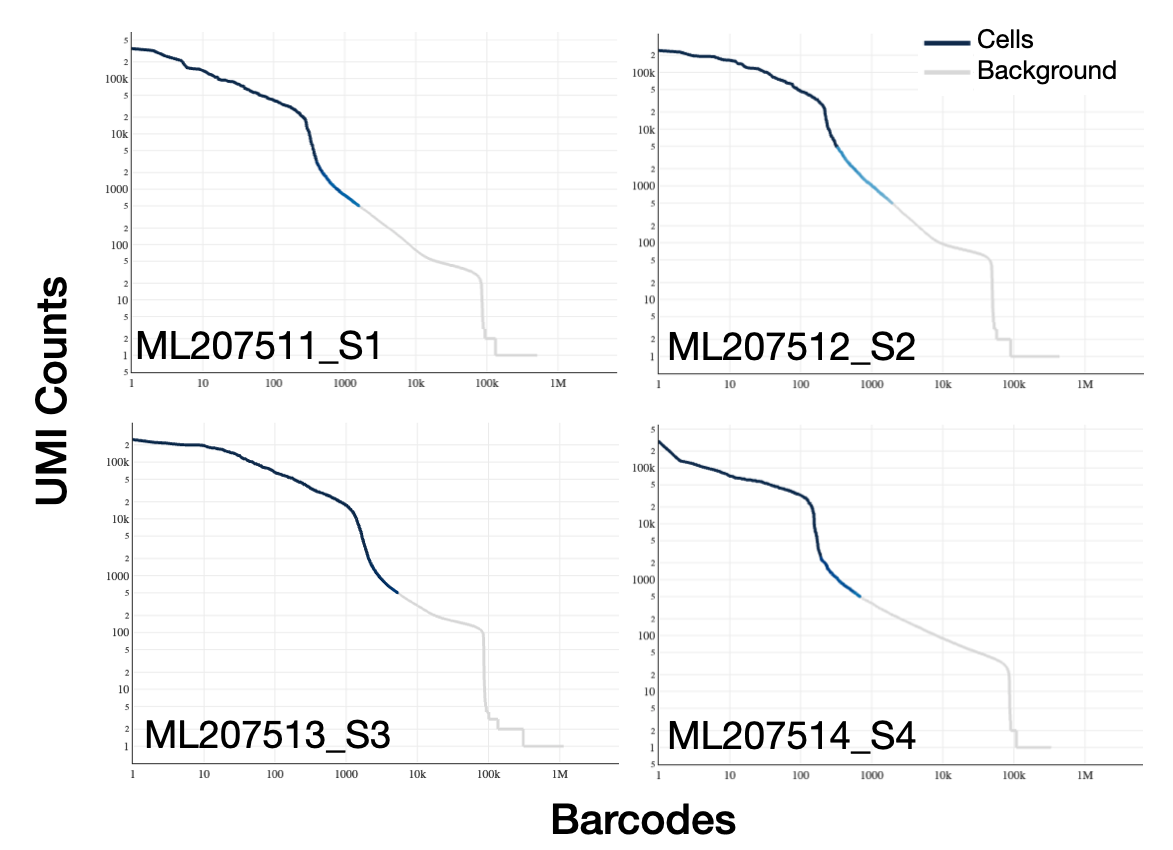
Knee plot cellranger on pilot #1 data (expected_cells=8k), the color of the line represents the local density of barcodes that are cell-associated.
However, in pilot #2, the knee plot shows that there are 15-20k cells with a UMI count >1000, suggesting there any many cells of intermediate quality.
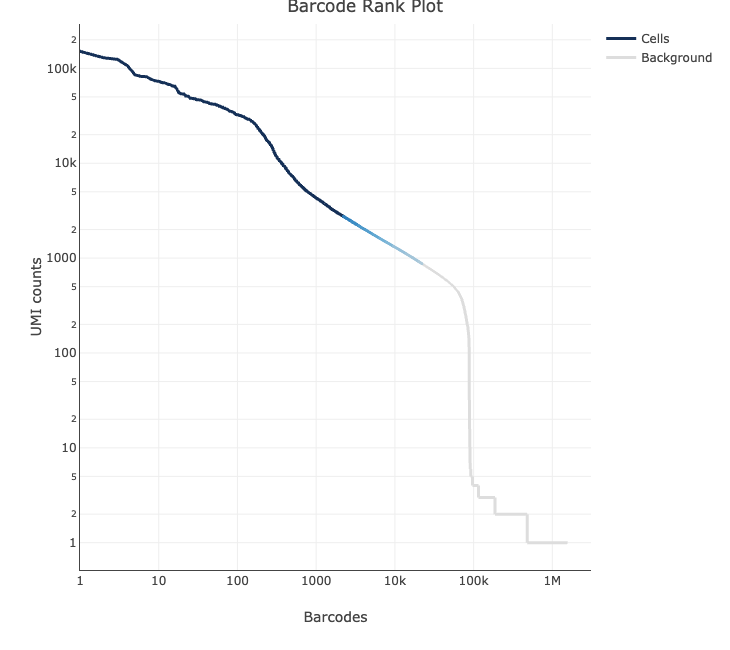
Knee plot cellranger on pilot #1 data (expected_cells=8k), the color of the line represents the local density of barcodes that are cell-associated.
Running EmptyDroplet on the data from the unfixed cells resulted in only 5.6k barcodes called as cells (609 for fixed cells), with only 29% (54% fixed) of reads from a cell associated barcode.
To capture these cells to assess by hand, instead of suggesting that cellranger look for 15k cells, I set forceCells=15k to ensure cellranger count returned the top 15k most likely cells. Summary statistics are shown here for the suggest/force runs and for the suggest mode run on the fixed cells. Even forcing EmptyDroplet to keep 15k cells, only 47% of reads were from cell associated barcodes, highlighting how extreme the ambient RNA problem just might be!
expected <- read.csv("/mnt/beegfs/mccarthy/scratch/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2_gex/01_cellranger/02_15kCExpect/cr-count_CB-scRNAv31-GEX-lib01/outs/metrics_summary.csv", header=TRUE)
expected$experiment <- "unfixed_suggest"
expected$Q30.Bases.in.Sample.Index <- NULL
fixed <- read.csv("/mnt/beegfs/mccarthy/scratch/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2_gex/01_cellranger/01_withoutExpectedCells/cr-count_CB-scRNAv31-GEX-lib02/outs/metrics_summary.csv", header=TRUE)
fixed$experiment <- "fixed_suggest"
fixed$Q30.Bases.in.Sample.Index <- NULL
forced <- read.csv("/mnt/beegfs/mccarthy/scratch/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot2_gex/01_cellrangerFORCE/cr-countFORCE_CB-scRNAv31-GEX-lib01/outs/metrics_summary.csv", header=TRUE)
forced$experiment <- "unfixed_force"
cr.summary <- rbind(fixed, expected, forced)
row.names(cr.summary) <- cr.summary$experiment
cr.summary$experiment <- NULL
cr.summary <- as.data.frame(t(cr.summary))
cr.summary fixed_suggest unfixed_suggest
Estimated.Number.of.Cells 609 5,695
Mean.Reads.per.Cell 1,105,060 124,575
Median.Genes.per.Cell 1,985 1,341
Number.of.Reads 672,981,585 709,456,102
Valid.Barcodes 97.2% 97.4%
Sequencing.Saturation 91.8% 62.0%
Q30.Bases.in.Barcode 96.2% 96.4%
Q30.Bases.in.RNA.Read 92.9% 93.3%
Q30.Bases.in.UMI 95.6% 96.0%
Reads.Mapped.to.Genome 94.0% 96.4%
Reads.Mapped.Confidently.to.Genome 84.1% 92.5%
Reads.Mapped.Confidently.to.Intergenic.Regions 8.4% 8.2%
Reads.Mapped.Confidently.to.Intronic.Regions 30.4% 51.2%
Reads.Mapped.Confidently.to.Exonic.Regions 45.3% 33.2%
Reads.Mapped.Confidently.to.Transcriptome 41.7% 29.7%
Reads.Mapped.Antisense.to.Gene 1.3% 2.5%
Fraction.Reads.in.Cells 53.7% 29.1%
Total.Genes.Detected 31,095 29,155
Median.UMI.Counts.per.Cell 3,773 2,150
unfixed_force
Estimated.Number.of.Cells 15,000
Mean.Reads.per.Cell 47,297
Median.Genes.per.Cell 864
Number.of.Reads 709,456,102
Valid.Barcodes 97.4%
Sequencing.Saturation 62.0%
Q30.Bases.in.Barcode 96.4%
Q30.Bases.in.RNA.Read 93.3%
Q30.Bases.in.UMI 96.0%
Reads.Mapped.to.Genome 96.4%
Reads.Mapped.Confidently.to.Genome 92.5%
Reads.Mapped.Confidently.to.Intergenic.Regions 8.2%
Reads.Mapped.Confidently.to.Intronic.Regions 51.2%
Reads.Mapped.Confidently.to.Exonic.Regions 33.2%
Reads.Mapped.Confidently.to.Transcriptome 29.7%
Reads.Mapped.Antisense.to.Gene 2.5%
Fraction.Reads.in.Cells 47.4%
Total.Genes.Detected 29,901
Median.UMI.Counts.per.Cell 1,497While we can force CellRanger to return more cells, this is likely to increase the amount of background noise in our dataset, ideally we want to use a different tool to call cells that is better at cell calling in the presence of high levels of ambient RNA.
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.0.4 (2021-02-15)
os Rocky Linux 8.4 (Green Obsidian)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2021-09-27
─ Packages ───────────────────────────────────────────────────────────────────
package * version date lib
assertthat 0.2.1 2019-03-21 [1]
backports 1.2.1 2020-12-09 [1]
beachmat 2.6.4 2020-12-20 [1]
Biobase * 2.50.0 2020-10-27 [1]
BiocGenerics * 0.36.1 2021-04-16 [1]
BiocParallel 1.24.1 2020-11-06 [1]
bitops 1.0-7 2021-04-24 [1]
broom 0.7.9 2021-07-27 [1]
bslib 0.2.5.1 2021-05-18 [1]
cachem 1.0.6 2021-08-19 [1]
callr 3.7.0 2021-04-20 [1]
cellranger 1.1.0 2016-07-27 [1]
cli 3.0.1 2021-07-17 [1]
colorspace 2.0-2 2021-06-24 [1]
crayon 1.4.1 2021-02-08 [1]
DBI 1.1.1 2021-01-15 [1]
dbplyr 2.1.1 2021-04-06 [1]
DelayedArray 0.16.3 2021-03-24 [1]
DelayedMatrixStats 1.12.3 2021-02-03 [1]
desc 1.3.0 2021-03-05 [1]
devtools 2.4.2 2021-06-07 [1]
digest 0.6.27 2020-10-24 [1]
dplyr * 1.0.7 2021-06-18 [1]
dqrng 0.3.0 2021-05-01 [1]
DropletUtils * 1.10.3 2021-02-02 [1]
edgeR 3.32.1 2021-01-14 [1]
ellipsis 0.3.2 2021-04-29 [1]
evaluate 0.14 2019-05-28 [1]
fansi 0.5.0 2021-05-25 [1]
fastmap 1.1.0 2021-01-25 [1]
forcats * 0.5.1 2021-01-27 [1]
fs 1.5.0 2020-07-31 [1]
generics 0.1.0 2020-10-31 [1]
GenomeInfoDb * 1.26.7 2021-04-08 [1]
GenomeInfoDbData 1.2.4 2020-11-10 [1]
GenomicRanges * 1.42.0 2020-10-27 [1]
ggplot2 * 3.3.5 2021-06-25 [1]
git2r 0.28.0 2021-01-10 [1]
glue 1.4.2 2020-08-27 [1]
gridExtra 2.3 2017-09-09 [1]
gtable 0.3.0 2019-03-25 [1]
haven 2.4.3 2021-08-04 [1]
HDF5Array 1.18.1 2021-02-04 [1]
hms 1.1.0 2021-05-17 [1]
htmltools 0.5.2 2021-08-25 [1]
httpuv 1.6.2 2021-08-18 [1]
httr 1.4.2 2020-07-20 [1]
IRanges * 2.24.1 2020-12-12 [1]
jcolors * 0.0.4 2020-11-23 [1]
jquerylib 0.1.4 2021-04-26 [1]
jsonlite 1.7.2 2020-12-09 [1]
knitr * 1.33 2021-04-24 [1]
later 1.3.0 2021-08-18 [1]
lattice 0.20-41 2020-04-02 [2]
lifecycle 1.0.0 2021-02-15 [1]
limma 3.46.0 2020-10-27 [1]
locfit 1.5-9.4 2020-03-25 [1]
lubridate 1.7.10 2021-02-26 [1]
magrittr 2.0.1 2020-11-17 [1]
Matrix * 1.3-4 2021-06-01 [1]
MatrixGenerics * 1.2.1 2021-01-30 [1]
matrixStats * 0.60.0 2021-07-26 [1]
memoise 2.0.0 2021-01-26 [1]
modelr 0.1.8 2020-05-19 [1]
munsell 0.5.0 2018-06-12 [1]
pillar 1.6.2 2021-07-29 [1]
pkgbuild 1.2.0 2020-12-15 [1]
pkgconfig 2.0.3 2019-09-22 [1]
pkgload 1.2.1 2021-04-06 [1]
prettyunits 1.1.1 2020-01-24 [1]
processx 3.5.2 2021-04-30 [1]
promises 1.2.0.1 2021-02-11 [1]
ps 1.6.0 2021-02-28 [1]
purrr * 0.3.4 2020-04-17 [1]
R.methodsS3 1.8.1 2020-08-26 [1]
R.oo 1.24.0 2020-08-26 [1]
R.utils 2.10.1 2020-08-26 [1]
R6 2.5.1 2021-08-19 [1]
Rcpp 1.0.7 2021-07-07 [1]
RCurl 1.98-1.4 2021-08-17 [1]
readr * 2.0.1 2021-08-10 [1]
readxl 1.3.1 2019-03-13 [1]
remotes 2.4.0 2021-06-02 [1]
reprex 2.0.1 2021-08-05 [1]
rhdf5 2.34.0 2020-10-27 [1]
rhdf5filters 1.2.1 2021-05-03 [1]
Rhdf5lib 1.12.1 2021-01-26 [1]
rlang 0.4.11 2021-04-30 [1]
rmarkdown 2.10 2021-08-06 [1]
rprojroot 2.0.2 2020-11-15 [1]
rstudioapi 0.13 2020-11-12 [1]
rvest 1.0.1 2021-07-26 [1]
S4Vectors * 0.28.1 2020-12-09 [1]
sass 0.4.0 2021-05-12 [1]
scales 1.1.1 2020-05-11 [1]
scuttle 1.0.4 2020-12-17 [1]
sessioninfo 1.1.1 2018-11-05 [1]
SingleCellExperiment * 1.12.0 2020-10-27 [1]
sparseMatrixStats 1.2.1 2021-02-02 [1]
stringi 1.7.3 2021-07-16 [1]
stringr * 1.4.0 2019-02-10 [1]
SummarizedExperiment * 1.20.0 2020-10-27 [1]
testthat 3.0.4 2021-07-01 [1]
tibble * 3.1.3 2021-07-23 [1]
tidyr * 1.1.3 2021-03-03 [1]
tidyselect 1.1.1 2021-04-30 [1]
tidyverse * 1.3.1 2021-04-15 [1]
tzdb 0.1.2 2021-07-20 [1]
usethis 2.0.1 2021-02-10 [1]
utf8 1.2.2 2021-07-24 [1]
vctrs 0.3.8 2021-04-29 [1]
viridis * 0.6.1 2021-05-11 [1]
viridisLite * 0.4.0 2021-04-13 [1]
withr 2.4.2 2021-04-18 [1]
workflowr 1.6.2 2020-04-30 [1]
xfun 0.25 2021-08-06 [1]
xml2 1.3.2 2020-04-23 [1]
XVector 0.30.0 2020-10-27 [1]
yaml 2.2.1 2020-02-01 [1]
zlibbioc 1.36.0 2020-10-27 [1]
source
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
Github (jaredhuling/jcolors@f200755)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.4)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.2)
Bioconductor
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
CRAN (R 4.0.4)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.2)
Bioconductor
[1] /mnt/mcfiles/cazodi/R/x86_64-pc-linux-gnu-library/4.0
[2] /opt/R/4.0.4/lib/R/library