2022-03-22_FANCM-manuscript-figures-3-dbcos
Ruqian Lyu
7/25/2021
Last updated: 2022-03-22
Checks: 5 1
Knit directory: yeln_2019_spermtyping/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the results is critical for reproducibility. To start using Git, open the Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full reproducibility benefits of using workflowr, please see ?wflow_start.
scCNV
From old analysis 2021-07-25_FANCM-manuscript-figures-3
Crossover interference
Find chromosomes with double crossovers
scCNV <-
readRDS(file = "~/Projects/rejy_2020_single-sperm-co-calling/output/outputR/analysisRDS/countsAll-settings4.3-scCNV-CO-counts_07-mar-2022.rds")
x <- c("mutant","mutant","wildtype","mutant",
"wildtype","wildtype")
xx <- c("Fancm-/-","Fancm-/-","Fancm+/+","Fancm-/-",
"Fancm+/+","Fancm+/+")cl <- makeCluster(3)
registerDoParallel(cl)
clusterEvalQ(cl, {
## set up each worker. Could also use clusterExport()
.libPaths("/mnt/beegfs/mccarthy/backed_up/general/rlyu/Software/Rlibs/4.1")
dyn.load("/mnt/mcfiles/rlyu/Software/miniconda3.4.7/envs/rlyubase/lib/libiconv.so.2")
dyn.load("/mnt/mcfiles/rlyu/Software/miniconda3.4.7/envs/rlyubase/lib/libicui18n.so.64")
NULL
})[[1]]
NULL
[[2]]
NULL
[[3]]
NULL##,
# .packages = c("BiocGenerics",
# "GenomicRanges",
# "SummarizedExperiment",
# "GenomeInfoDb")
chrCOs_CNV <- foreach(chr = paste0("chr",1:19),.combine = "rbind",
.packages = c("GenomeInfoDb","SummarizedExperiment")) %dopar% {
tmp <- scCNV[seqnames(scCNV)==chr,]
data.frame(barcode = names(colSums(as.matrix(assay(tmp)))),
COs = colSums( as.matrix(assay(tmp))),
Chrom = chr)
}
dbCO_barcodes <- chrCOs_CNV[chrCOs_CNV$COs==2,] Double crossover distribution
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.8, 0.8))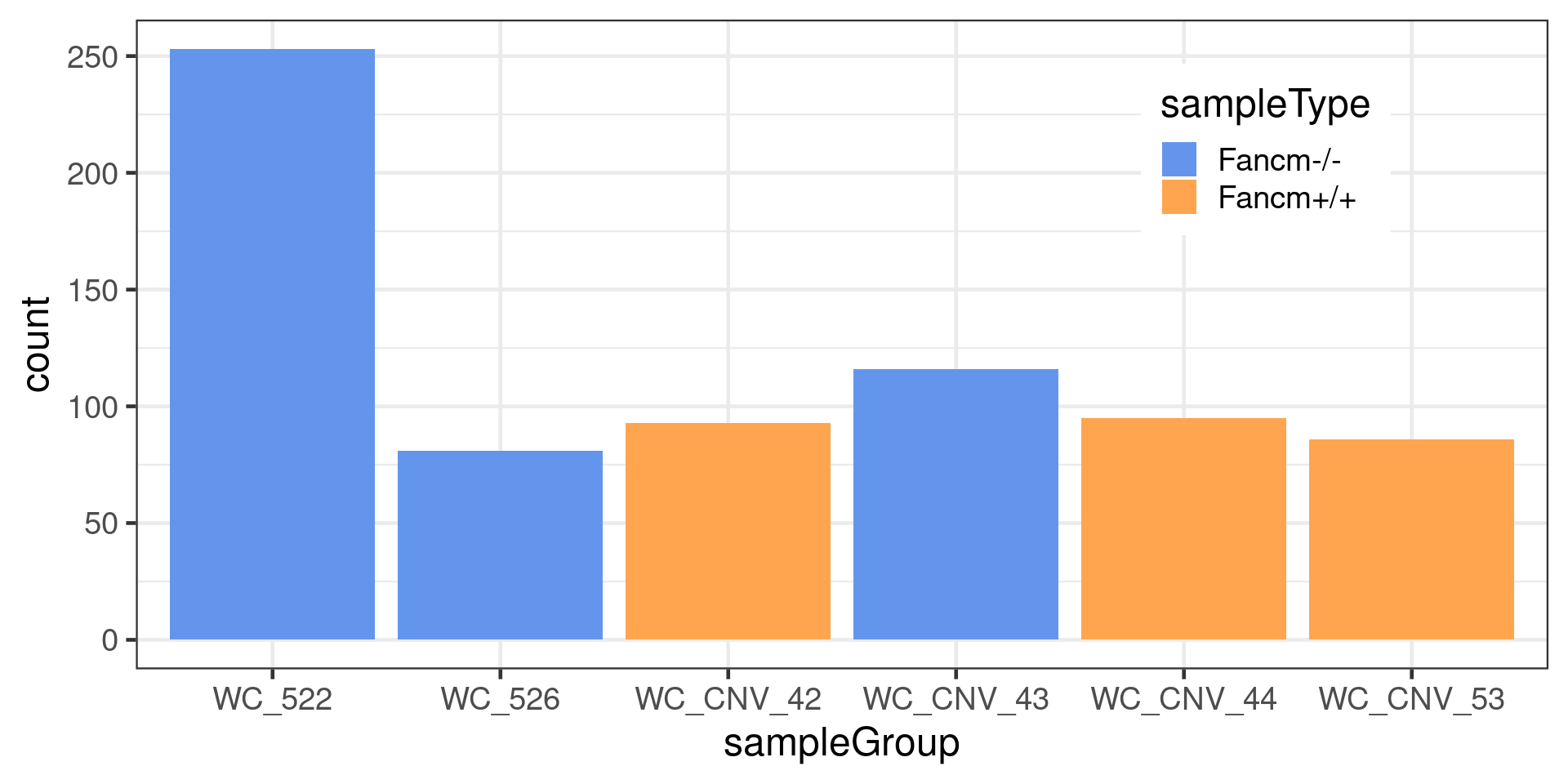
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.8, 0.8))+guides(fill =FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.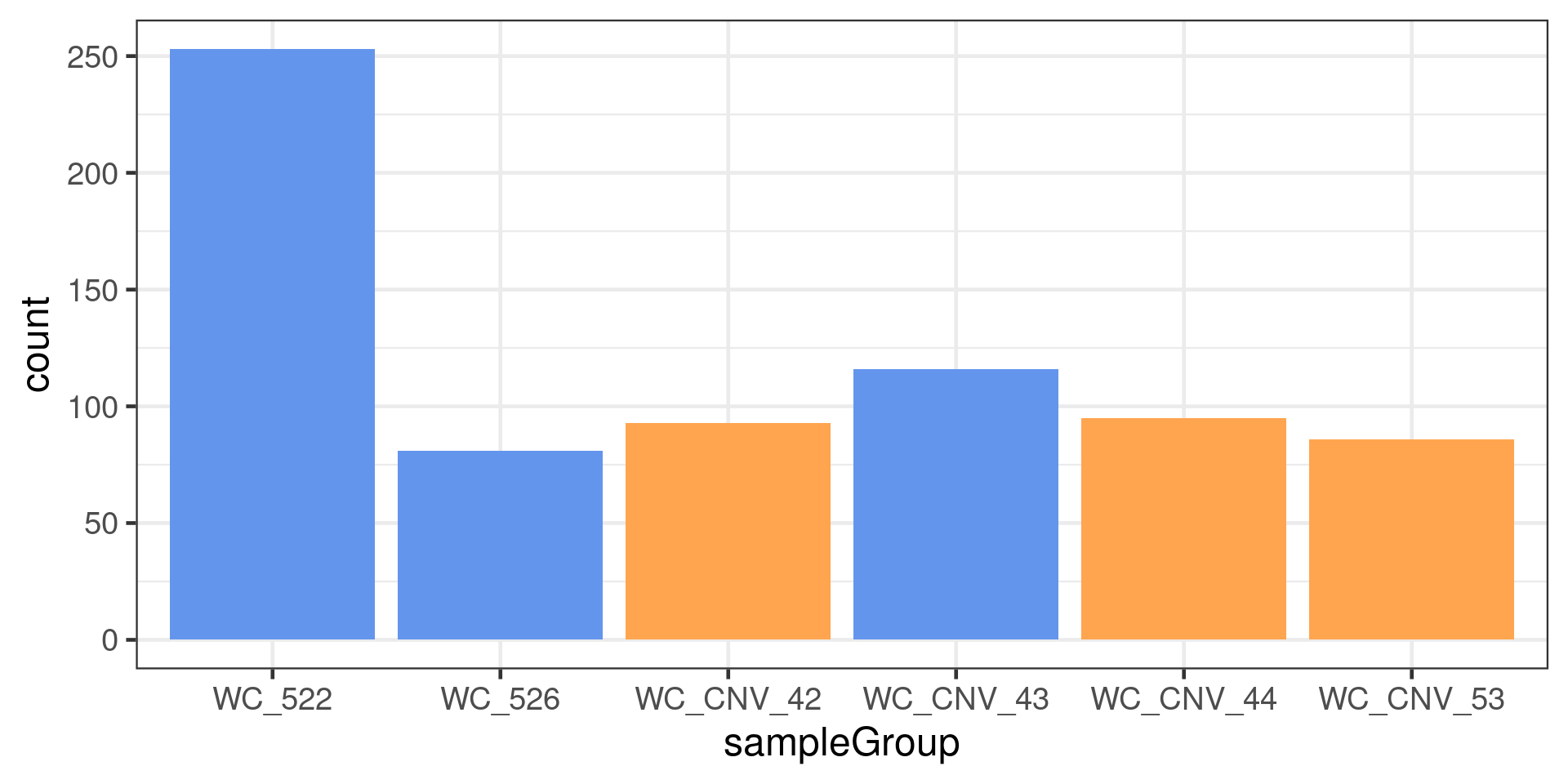
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleType,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+theme(legend.position = c(0.8, 0.8))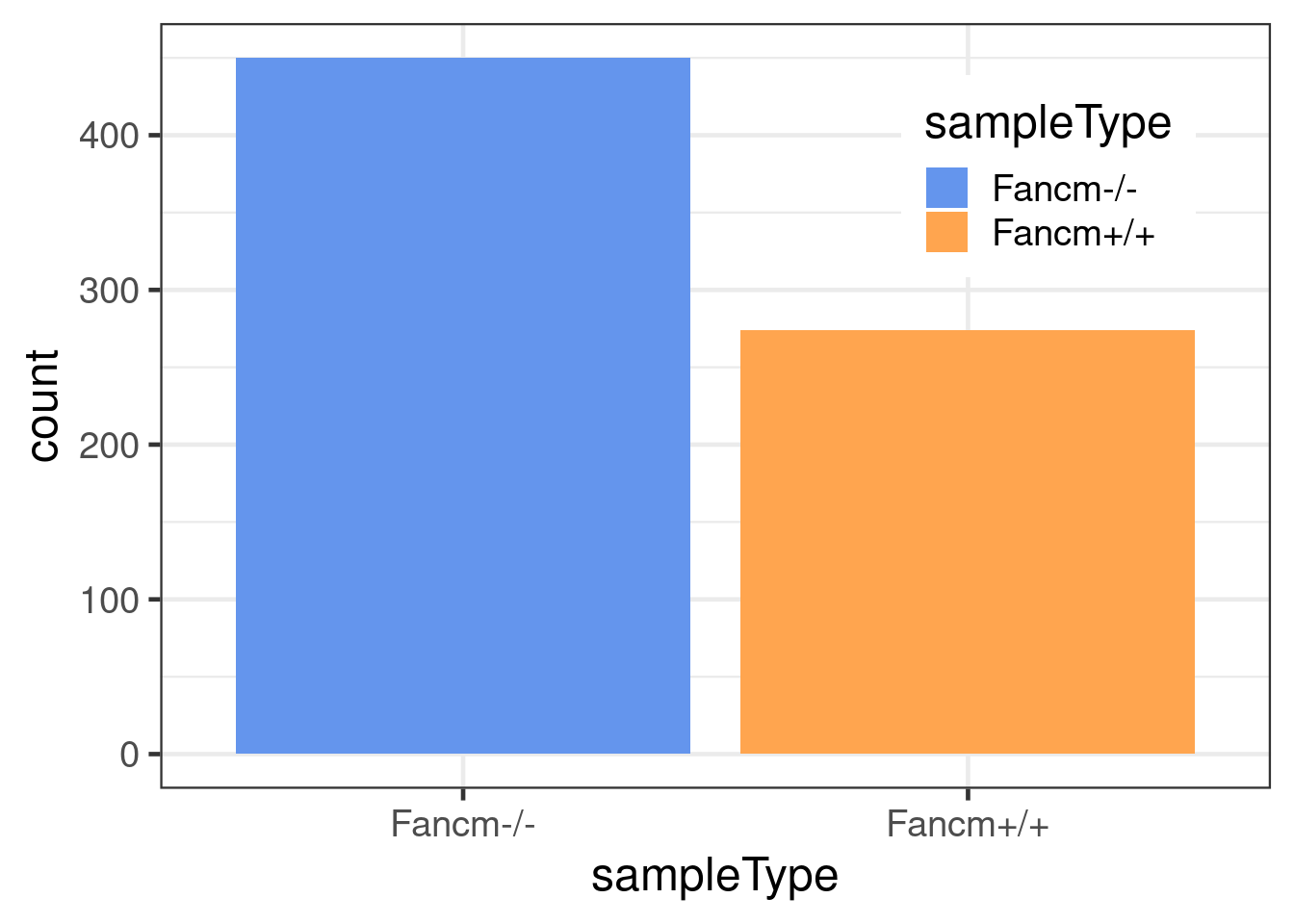
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleType,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+theme(legend.position = c(0.8, 0.8))+
guides(fill = FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.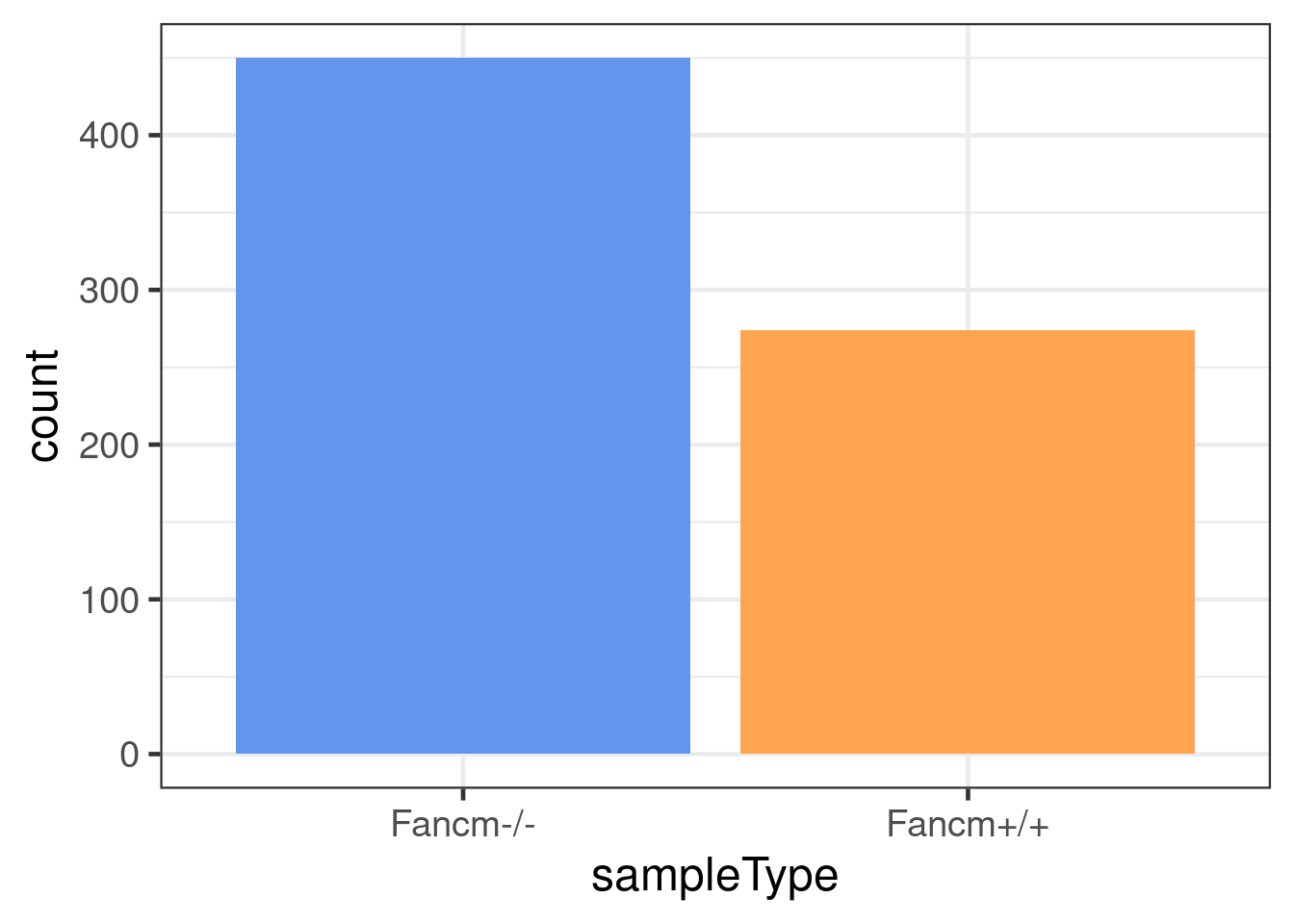
scCNV rate of double crossover chromosomes
There might be more sperms from mutant individuals, so we calculate the mean double crossover rate.
nSperms <- data.frame(nSperms = table(scCNV$sampleGroup))
colnames(nSperms) <- c("sampleGroup","Freq")
nSperms <- nSperms %>% dplyr::mutate(sampleType = as.character(plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)))The number of chromosomes with double crossovers normalised by the number of sperms per sample
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% dplyr::group_by(sampleGroup) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$Freq),
sampleType = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$sampleType)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.8, 0.8))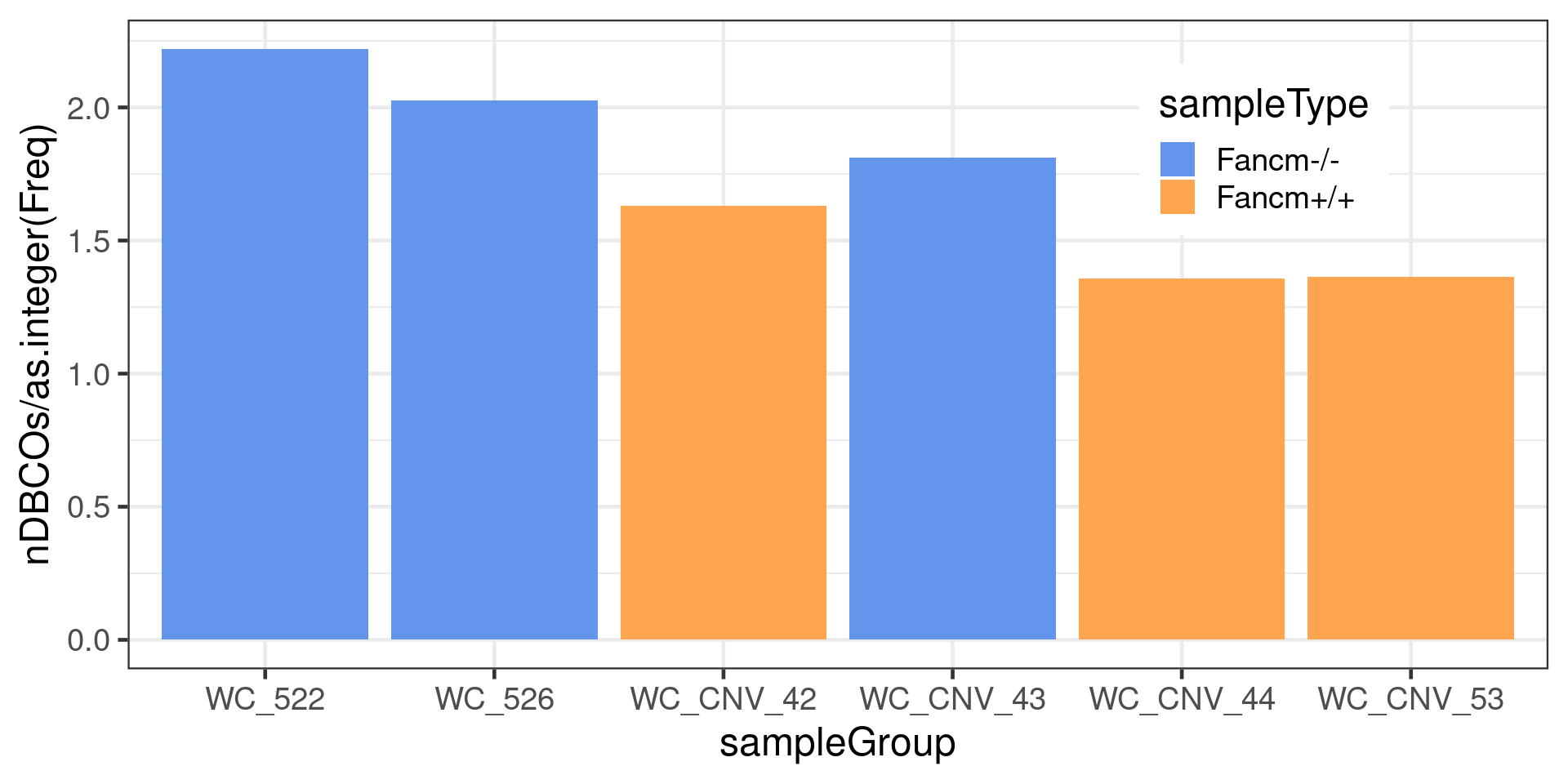
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% dplyr::group_by(sampleGroup) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$Freq),
sampleType = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$sampleType)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.8, 0.8))+guides(fill=FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.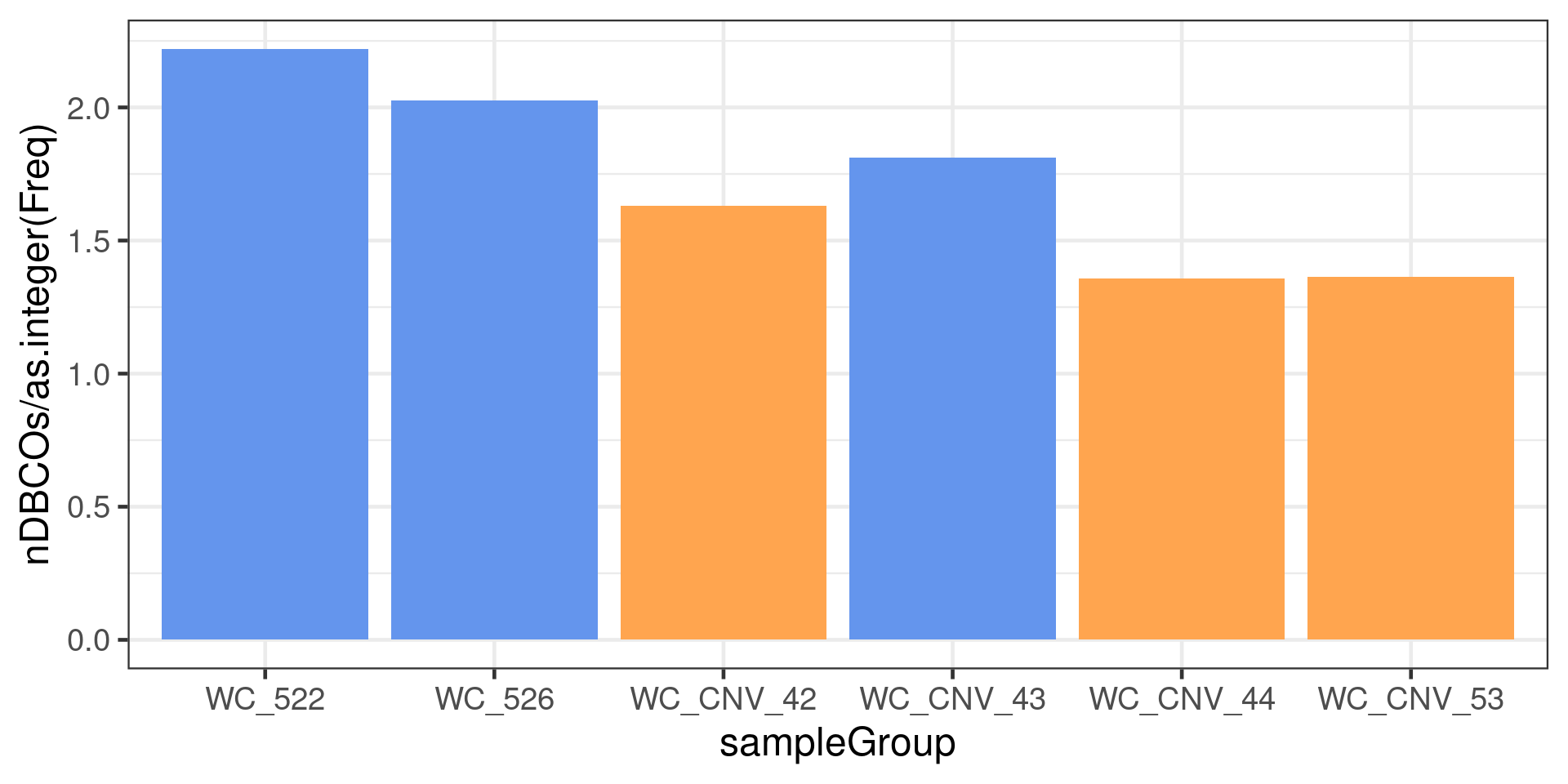
The number of chromosomes with double crossovers normalised by the number of sperms per genotype
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% dplyr::group_by(sampleGroup) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$Freq),
sampleType = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$sampleType)) %>%
dplyr::group_by(sampleType) %>% dplyr::summarise(nDBCOs = sum(.data$nDBCOs),
Freq = sum(as.integer(.data$Freq))) %>%
ggplot() +
geom_bar(mapping = aes(x = sampleType, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.74, 0.8))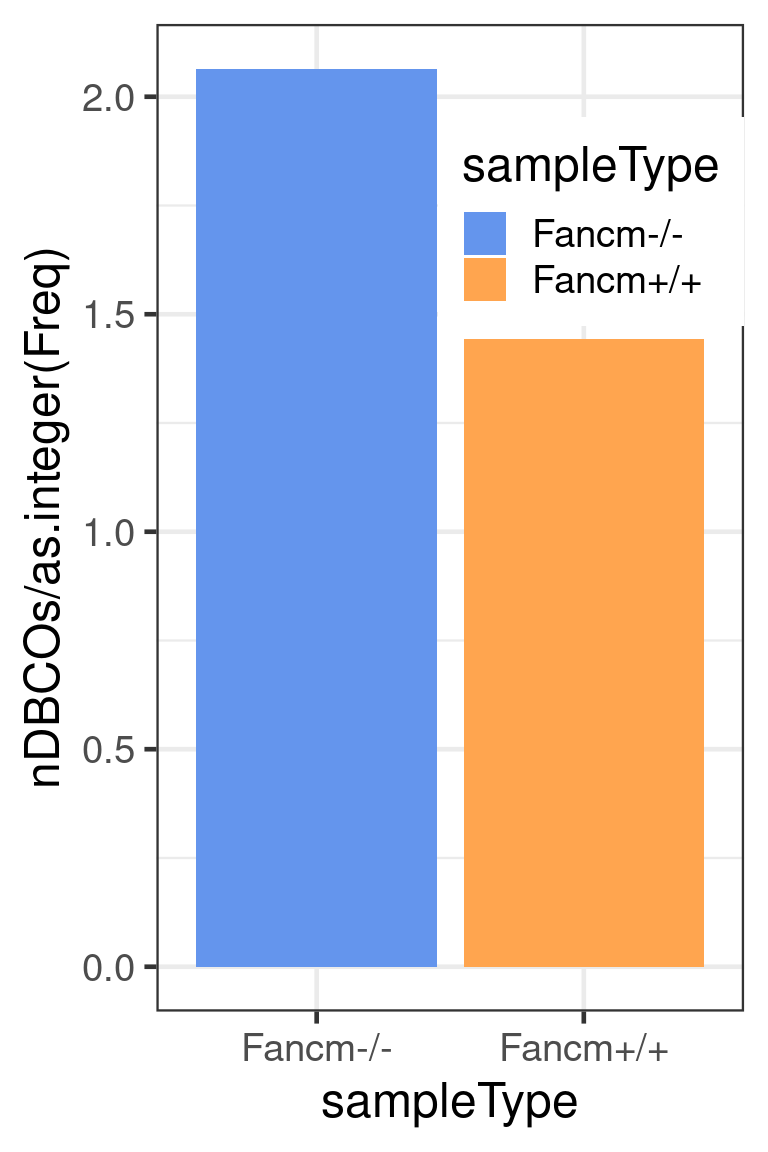
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx)) %>% dplyr::group_by(sampleGroup) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$Freq),
sampleType = plyr::mapvalues(sampleGroup,
from = nSperms$sampleGroup,
to = nSperms$sampleType)) %>%
dplyr::group_by(sampleType) %>% dplyr::summarise(nDBCOs = sum(.data$nDBCOs),
Freq = sum(as.integer(.data$Freq))) %>%
ggplot() +
geom_bar(mapping = aes(x = sampleType, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.74, 0.8))+guides(fill=FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.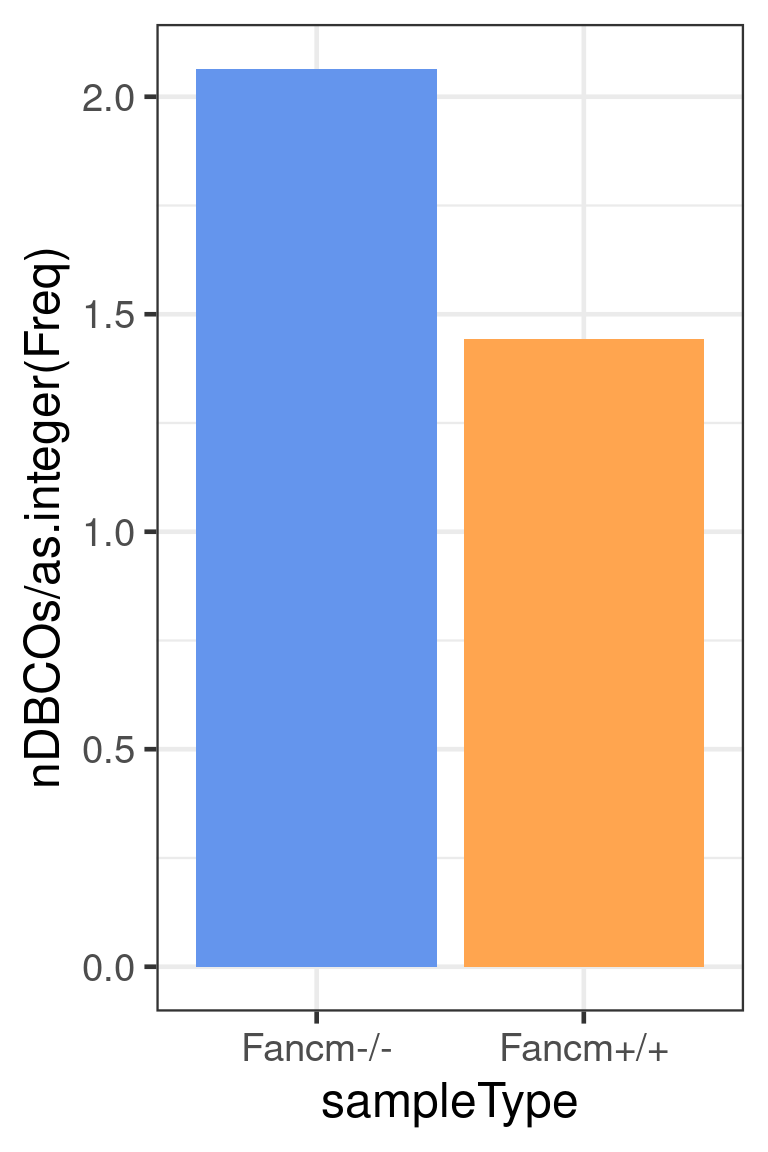
Inter-crossover distances
double_CO_intervals_scCNV <- foreach(bc = unique(dbCO_barcodes$barcode),.combine = "rbind",
.packages = c("BiocGenerics",
"GenomicRanges","SummarizedExperiment")) %dopar%
{
dd <- data.frame(start_pos =
start(comapr::getCellCORange(scCNV,
cellBarcode =bc)),
end_pos = end(comapr::getCellCORange(scCNV,
cellBarcode =bc)),
Chrom = as.character(seqnames(comapr::getCellCORange(scCNV,
cellBarcode = bc))),
barcode = bc)
keepChrs <- names( table(dd$Chrom)[table(dd$Chrom)==2])
dd[dd$Chrom%in% keepChrs,]
}double_CO_intervals_scCNV$coID <- paste0(double_CO_intervals_scCNV$Chrom,"_",
double_CO_intervals_scCNV$barcode)
double_CO_intervals_scCNV <- double_CO_intervals_scCNV %>%
dplyr::mutate(sampleGroup = colData(scCNV)[barcode,"sampleGroup"]) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =xx))
secondCO <- double_CO_intervals_scCNV[duplicated(double_CO_intervals_scCNV$coID),]
firstCO <- double_CO_intervals_scCNV[!duplicated(double_CO_intervals_scCNV$coID),]
double_CO_CNV_wide <- merge(firstCO,secondCO,
by = c("coID","sampleGroup","sampleType",
"barcode","Chrom"))double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist))+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.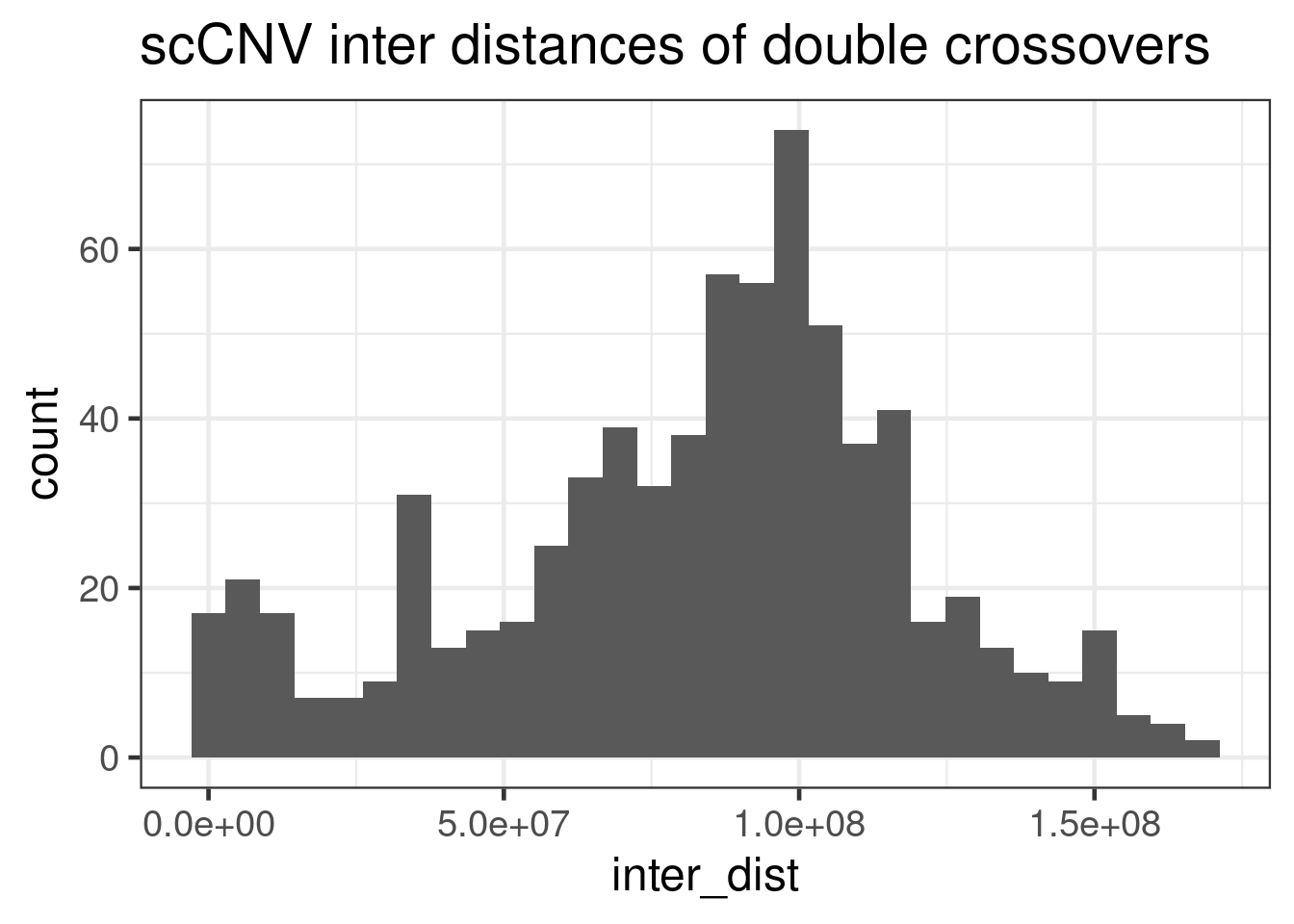
double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleType))+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.74, 0.8),
legend.title = element_blank())+xlab("Nucleotide distance between double crossovers")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.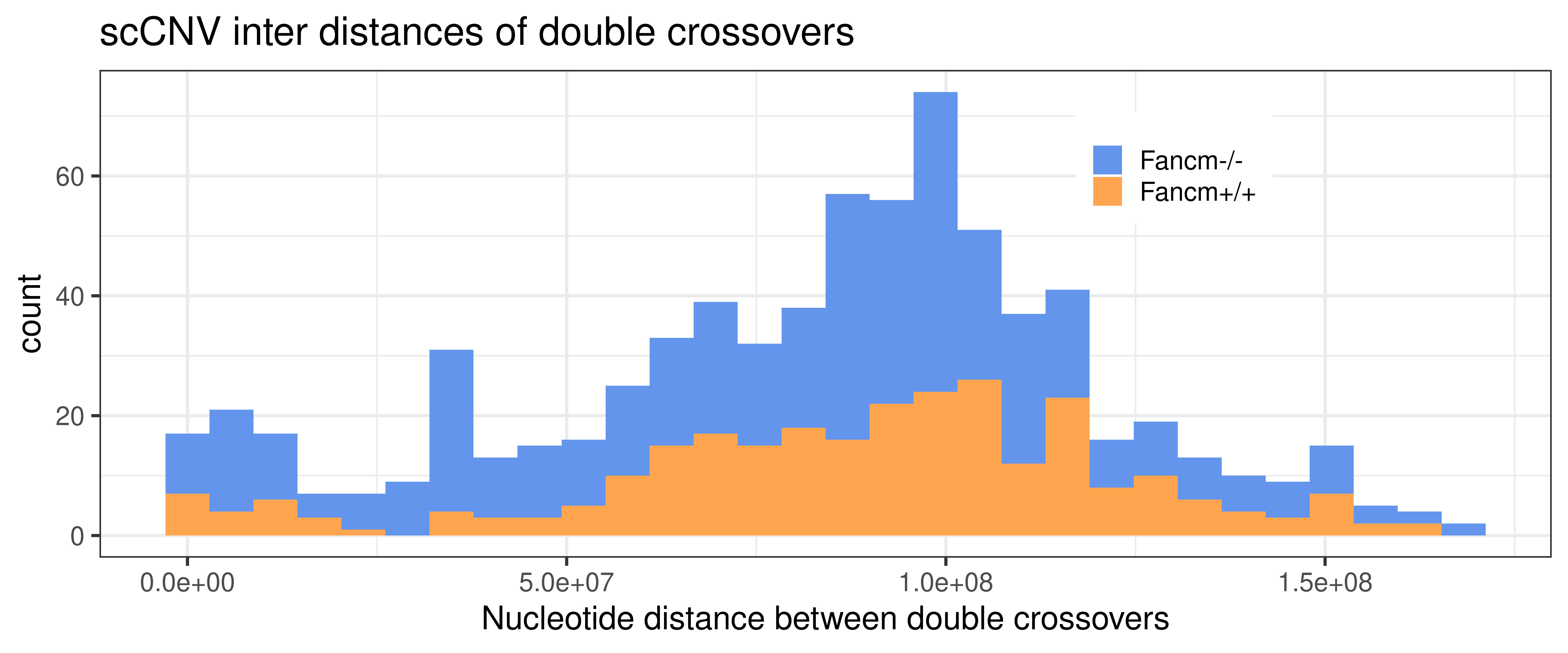
double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleType))+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"),
labels = c("Fancm-/-"= expression(italic(Fancm^Delta*""^"2/"*""^Delta*""^"2")),
"Fancm+/+" = expression(italic("Fancm"^"+/+" ))))+
theme(legend.position = c(0.74, 0.8))+xlab("Nucleotide distance between double crossovers")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.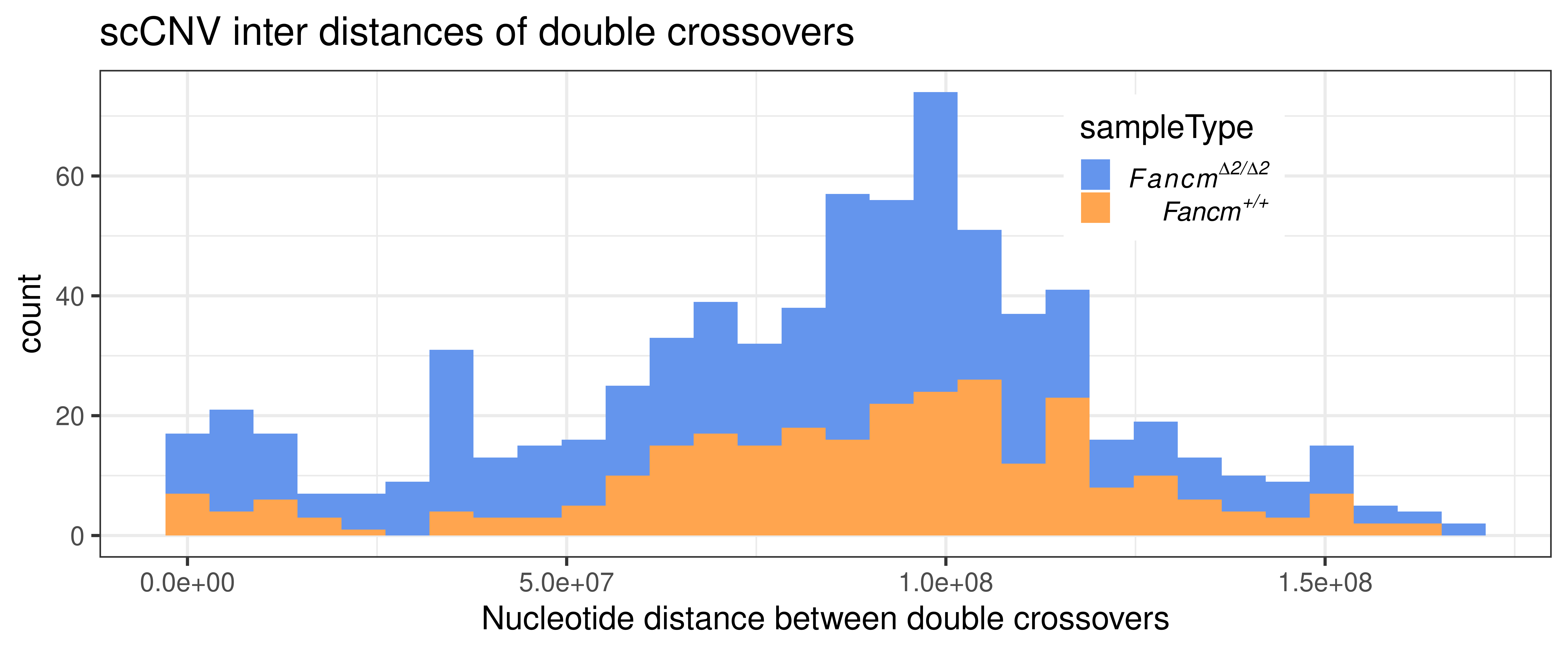
p_double_CO_CNV_wide <- double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleType))+theme_bw(base_size = 24) +
ggtitle("F1 single-sperm sequencing ")+ scale_x_continuous(labels = scales::unit_format(unit = "M",
scale = 1e-06))+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"),
labels = c("Fancm-/-"= expression(italic(Fancm^Delta*""^"2/"*""^Delta*""^"2")),
"Fancm+/+" = expression(italic("Fancm"^"+/+" ))))+xlab("Nucleotide distance between double crossovers")+
ylab("Count")+theme(legend.position = c(0.78, 0.85),legend.text.align = 0,legend.text = element_text(size = 25),
legend.background = element_blank(),legend.title = element_blank(),
axis.text = element_text(size = 25),
axis.title = element_text(size = 25))
p_double_CO_CNV_wide`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.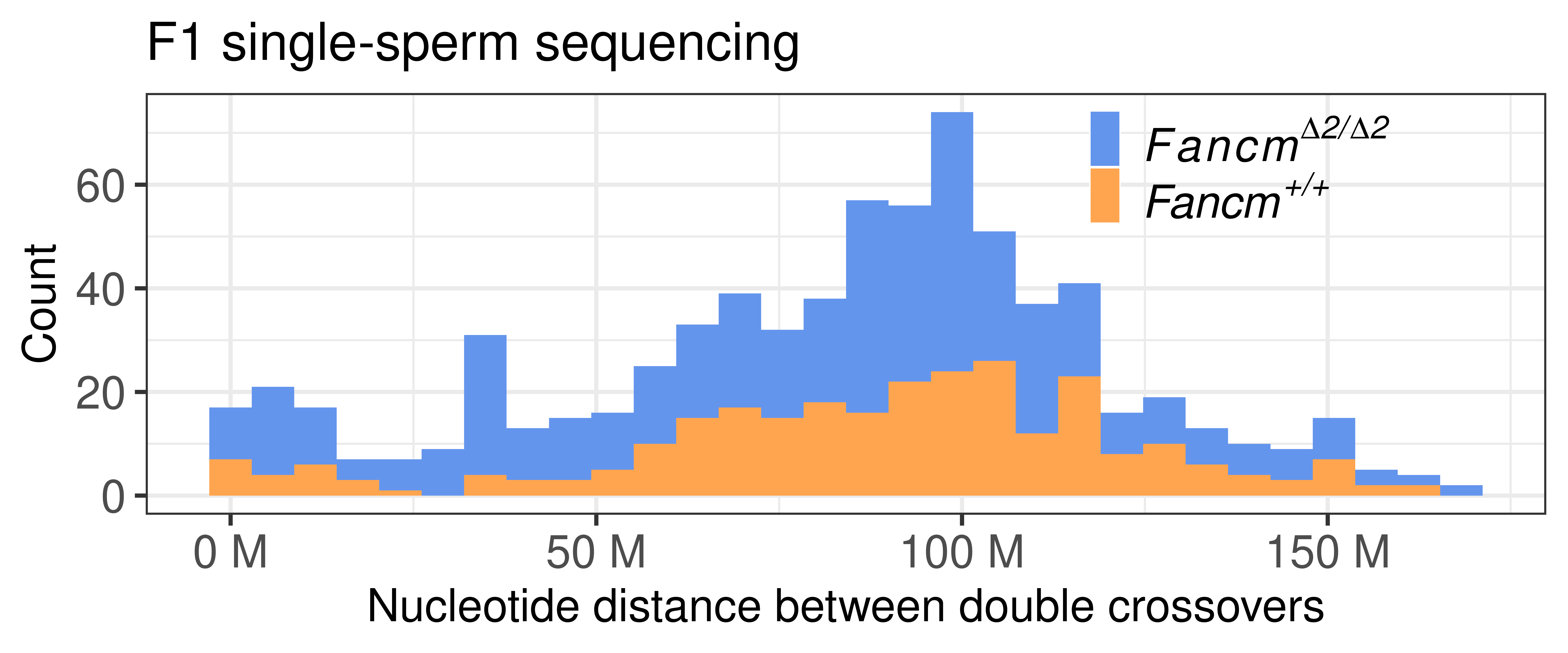
ggsave(filename = "output/outputR/analysisRDS/figures/double_CO_CNV_wide-hist.png",
plot = p_double_CO_CNV_wide,
dpi = 300, width = 12)Number of double crossovers per group scCNV
table(double_CO_CNV_wide$sampleType)
Fancm-/- Fancm+/+
453 276 double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_density(mapping = aes(inter_dist,
fill = sampleType),alpha=0.3)+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.78, 0.8),
legend.title = element_blank())+xlab("Nucleotide distance between double crossovers")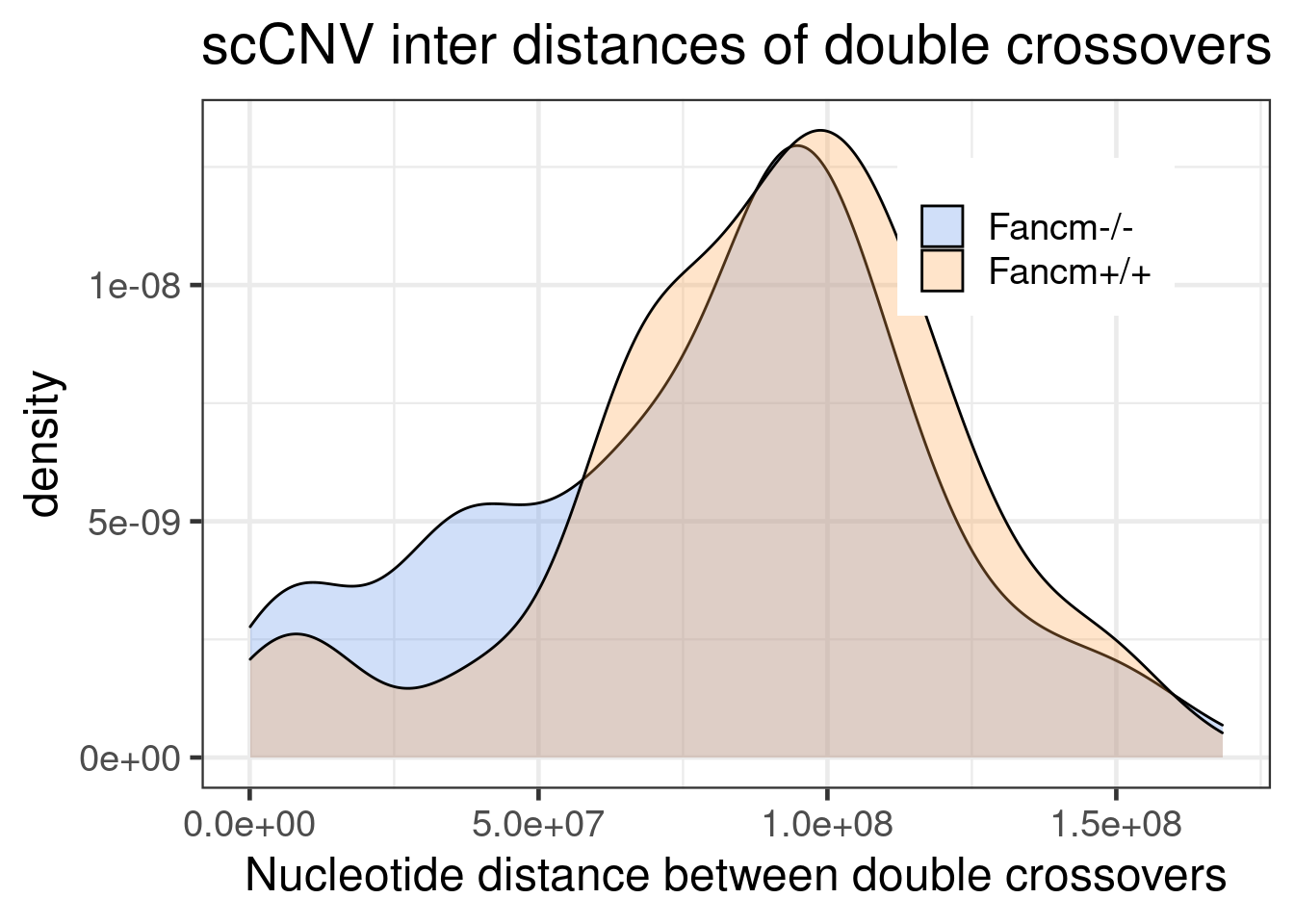
double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_density(mapping = aes(inter_dist,
fill = sampleType),alpha=0.3)+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
scale_fill_manual(values = c("Fancm-/-" = "cornflowerblue",
"Fancm+/+" = "tan1"))+
theme(legend.position = c(0.78, 0.8))+guides(fill = FALSE)+
xlab("Nucleotide distance between double crossovers")Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.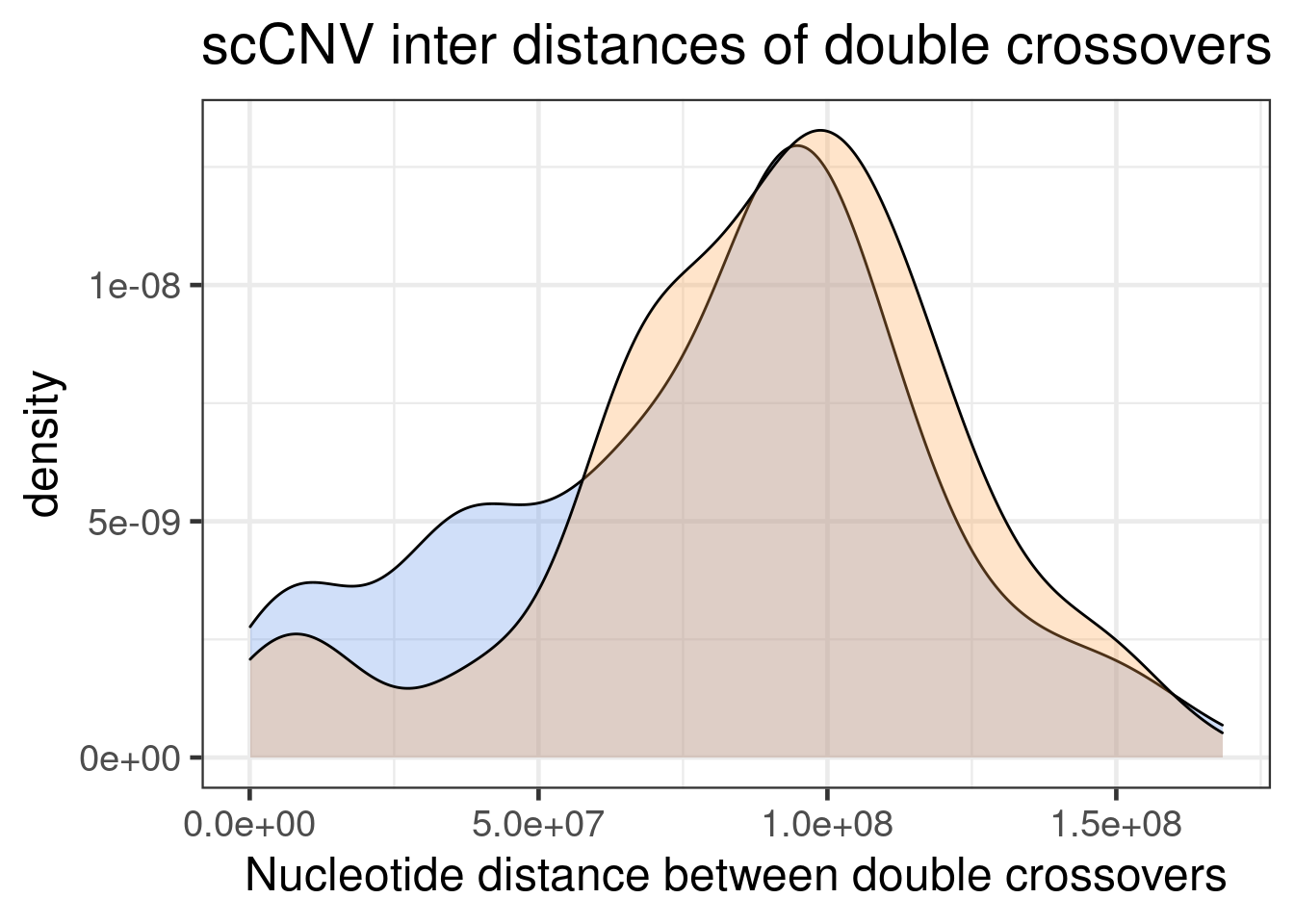
mt_dist <- double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter( sampleType == "Fancm-/-") %>% dplyr::select(inter_dist)
wt_dist <- double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter( sampleType == "Fancm+/+") %>% dplyr::select(inter_dist)
wilcox.test(mt_dist$inter_dist,wt_dist$inter_dist,alternative = "less")
Wilcoxon rank sum test with continuity correction
data: mt_dist$inter_dist and wt_dist$inter_dist
W = 53520, p-value = 0.000555
alternative hypothesis: true location shift is less than 0double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleGroup))+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.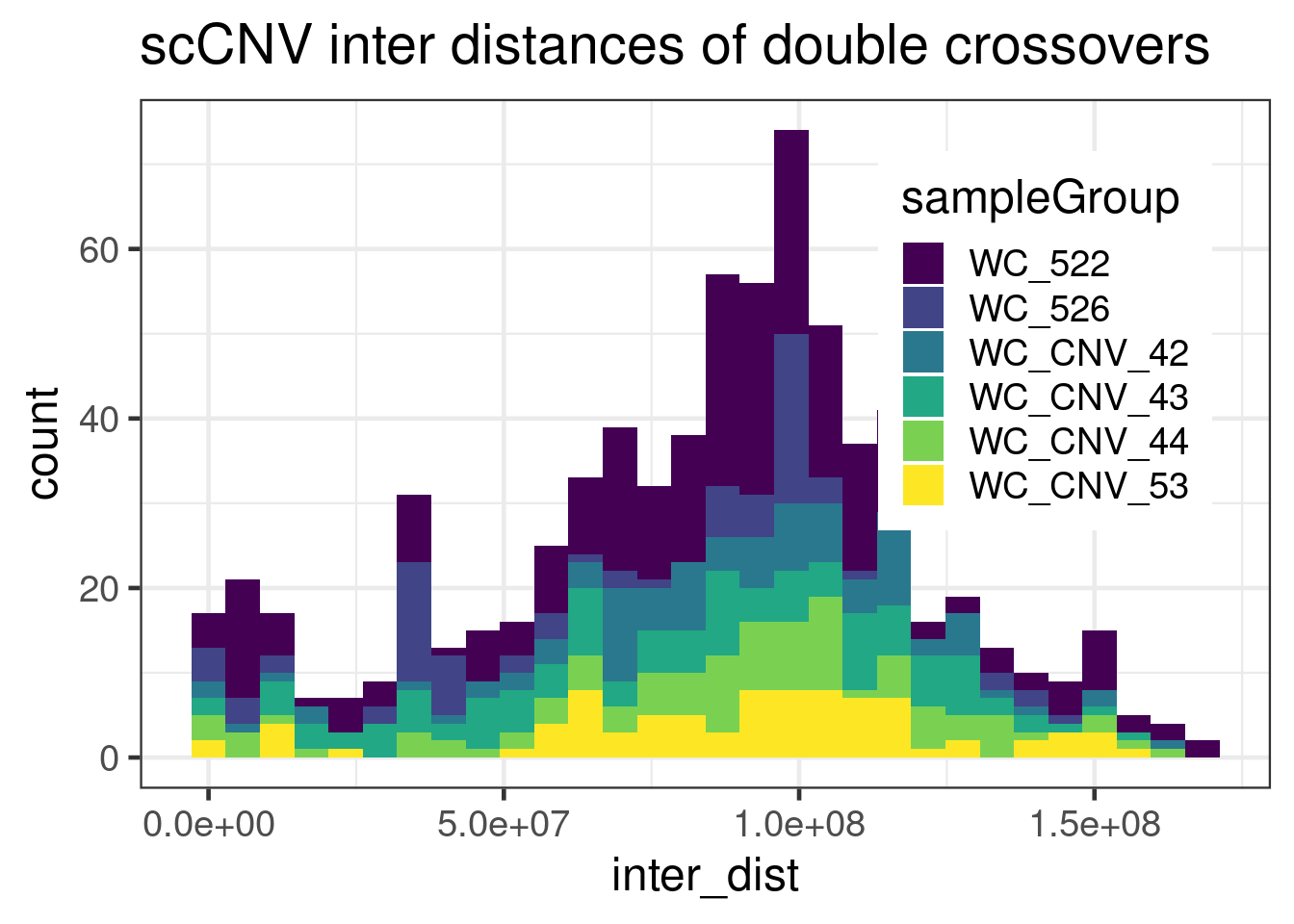
double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleGroup))+theme_bw(base_size = 18) +
ggtitle("scCNV inter distances of double crossovers ")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()+guides(fill = FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.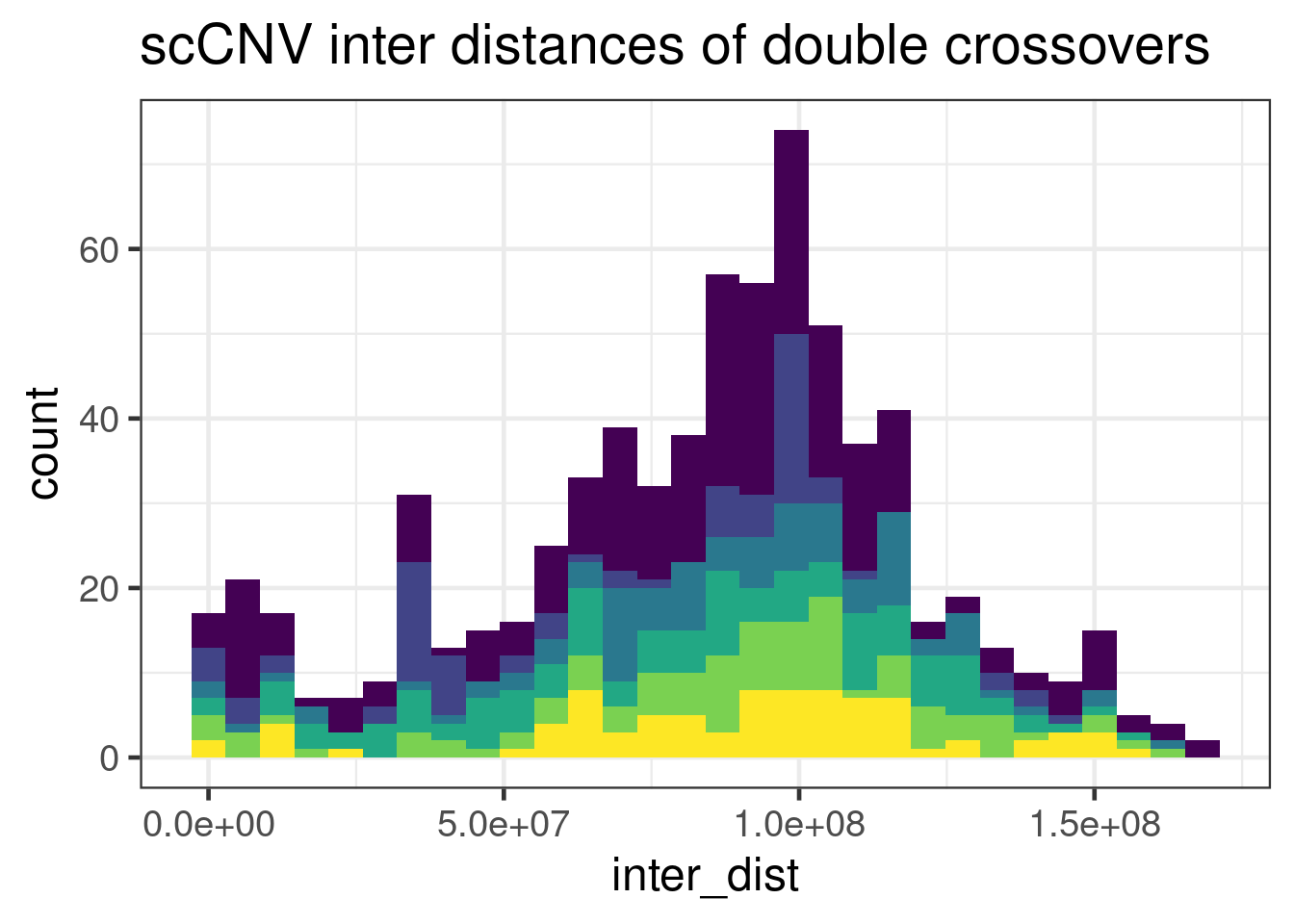
crossover interference by permuting the crossover positions across the chromosomes
permuteOnce <- function(firstCOs, secondCOs, chr){
firstCOs <- firstCOs[firstCOs$Chrom ==chr,]
secondCOs <- secondCOs[secondCOs$Chrom ==chr,]
allCOs <- seq(nrow(firstCOs)*2)
permCOs <- sample(allCOs)
tmp <- rbind(firstCOs,secondCOs)
tmp <- tmp[permCOs,]
tmp$coID <- rep(firstCOs$coID,each=2)
#tmp
tmpFirst <- tmp[duplicated(tmp$coID),]
tmpSecond <- tmp[!duplicated(tmp$coID),]
double_CO_pemute_wide <- merge(tmpFirst,tmpSecond,
by =c("coID","Chrom"))
double_CO_pemute_wide <- double_CO_pemute_wide[,c("coID","Chrom","start_pos.x",
"end_pos.x","start_pos.y",
"end_pos.y")]
double_CO_pemute_wide
}Fancm -/- sperms
Permute 1000 times for mutant/wildtypes cells
B <- 1000
set.seed(2021)
mutant_scCNV_permute <- foreach(b = seq(B), .combine = "rbind") %dopar% {
pm <- lapply(unique(firstCO[firstCO$sampleType=="Fancm-/-","Chrom"]),
function(chr){
permuteOnce(firstCOs = firstCO[firstCO$sampleType=="Fancm-/-",],
secondCOs = secondCO[secondCO$sampleType=="Fancm-/-",],
chr = chr)
})
pm <- do.call(rbind,pm)
}
#head(mutant_permute)wt_scCNV_permute <- foreach(b = seq(B), .combine = "rbind") %dopar% {
pm <- lapply(unique(firstCO[firstCO$sampleType=="Fancm+/+","Chrom"]),
function(chr){
permuteOnce(firstCOs = firstCO[firstCO$sampleType=="Fancm+/+",],
secondCOs = secondCO[secondCO$sampleType=="Fancm+/+",],
chr = chr)
})
pm <- do.call(rbind,pm)
}Plot null distribution of inter-crossover distance versus observed inter-crossover distances
wt_scCNV_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes( abs(inter_dist)))+theme_bw(base_size = 18) +
ggtitle("F1 single-sperm sequencing")+xlab("null distribution of inter distances of double crossovers for Fancm+/+ cells")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.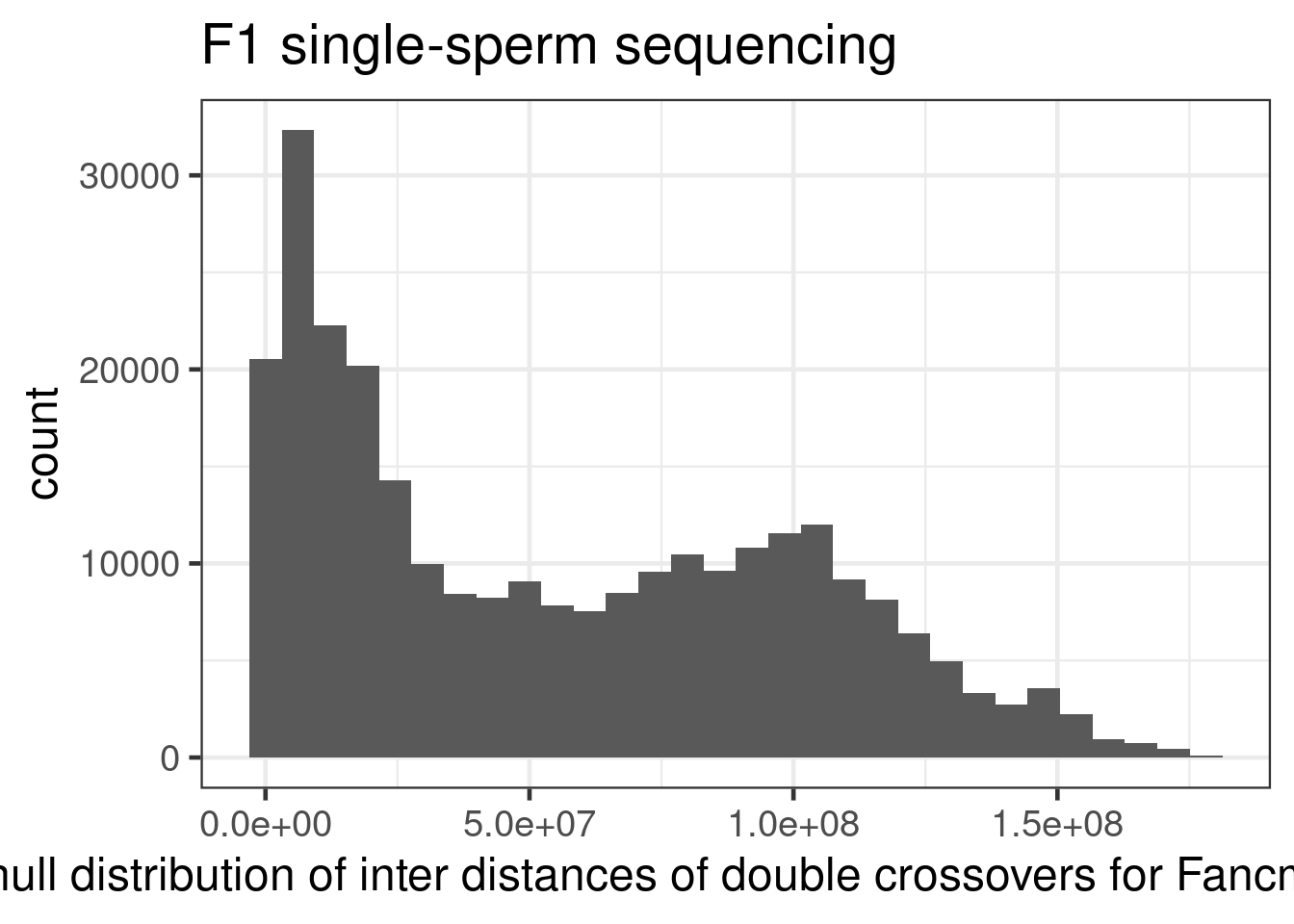
mutant_scCNV_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(abs(inter_dist)))+theme_bw(base_size = 18) +
ggtitle("F1 single-sperm sequencing")+xlab("null distribution of inter distances of double crossovers for Fancm-/- cells")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.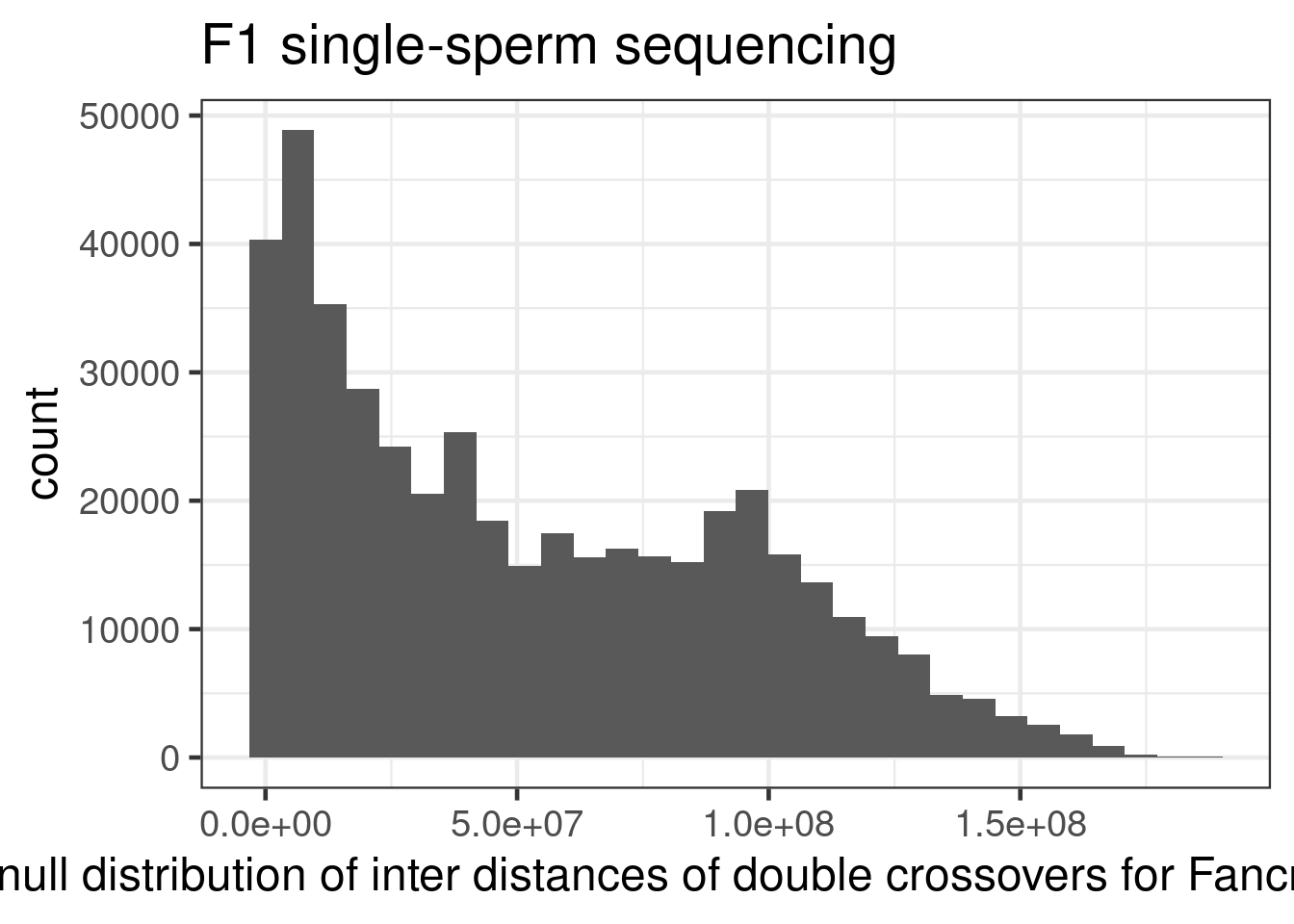
Observed versus the null for mutant cells
mutant_scCNV_permute <- mutant_scCNV_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>% dplyr::mutate(source = "null")
mutant_double_CO_CNV_wide <- double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter(sampleType== "Fancm-/-") %>%
dplyr::mutate(source = "observed") %>%
dplyr::select(colnames(mutant_scCNV_permute))p_mt_null_observed <- rbind(mutant_scCNV_permute, mutant_double_CO_CNV_wide) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.3)+theme_bw(base_size = 18) +
ggtitle("F1 single-sperm sequencing ")+xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.7, 0.86),
legend.title = element_blank(),
legend.background = element_blank())+
scale_fill_manual(labels = c("null" = "Null distribution",
"observed" = "observed (Fancm-/-)"),
values = c("null" = "grey",
"observed" = "cornflowerblue"))
p_mt_null_observed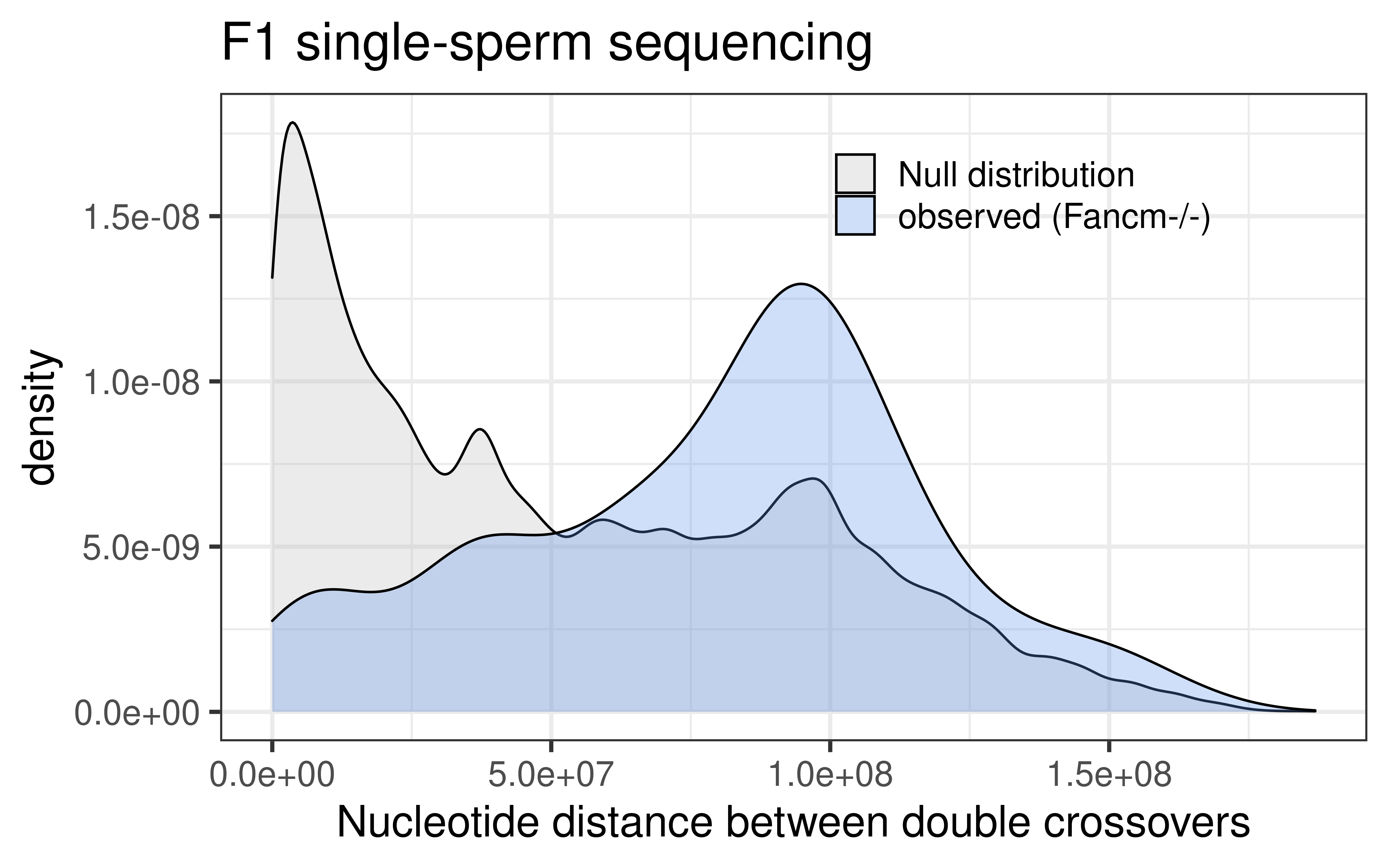
Observed versus the null for wildtype
wt_scCNV_permute <- wt_scCNV_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>% dplyr::mutate(source = "null")
wt_double_CO_CNV_wide <- double_CO_CNV_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter(sampleType== "Fancm+/+") %>%
dplyr::mutate(source = "observed") %>%
dplyr::select(colnames(wt_scCNV_permute))p_wt_null_observed <- rbind(wt_scCNV_permute, wt_double_CO_CNV_wide) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.2)+theme_bw(base_size = 18) +
ggtitle("F1 single-sperm sequencing ")+xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.75, 0.92),
legend.title = element_blank(),
legend.background = element_blank())+
scale_fill_manual(labels = c("null" = "Null distribution",
"observed" = "observed (Fancm+/+) "),
values = c("null" = "grey",
"observed" = "tan1"))
p_wt_null_observed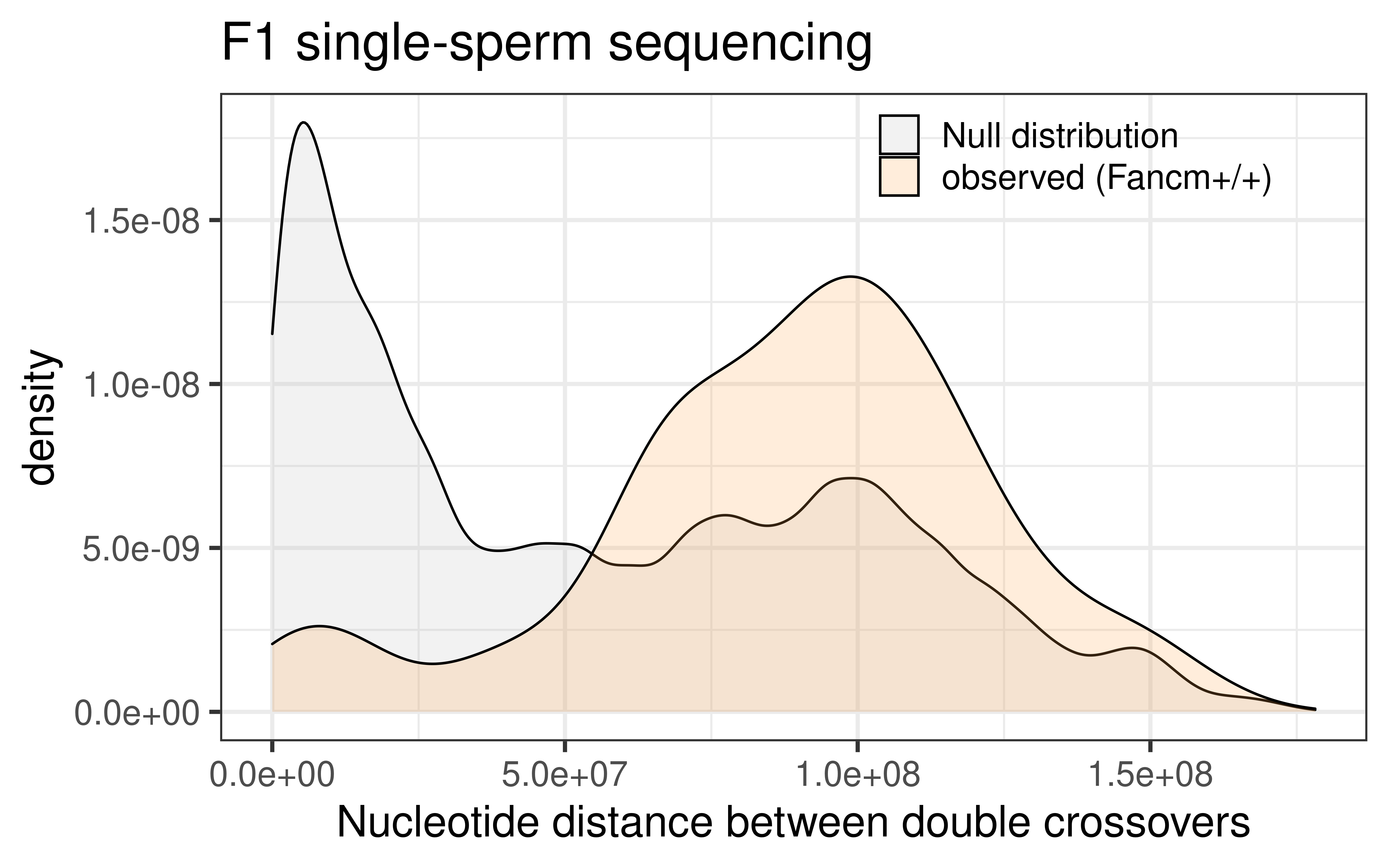
wt_scCNV_panel <- rbind(wt_scCNV_permute, wt_double_CO_CNV_wide)
wt_scCNV_panel$source <- plyr::mapvalues(wt_scCNV_panel$source,from =c("null","observed"),
to = c("null","wt_observed"))
wt_scCNV_panel$sampleType <- "wt"
mt_scCNV_panel <- rbind(mutant_scCNV_permute, mutant_double_CO_CNV_wide)
mt_scCNV_panel$source <- plyr::mapvalues(mt_scCNV_panel$source,from =c("null","observed"),
to = c("null","mt_observed"))
mt_scCNV_panel$sampleType <- "mt"
p_dbco_dist_scCNV_twopanels <- dbco_dist_scCNV <- rbind(mt_scCNV_panel,wt_scCNV_panel) %>% mutate(sampleType = factor(sampleType,levels = c("wt","mt"))) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.2)+
scale_x_continuous(labels = scales::unit_format(unit = "M",
scale = 1e-06))+
theme_bw(base_size = 20) +
# ggtitle("F1 single-sperm sequencing ")+
xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.8, 0.87),
legend.title = element_blank(),
legend.background = element_blank(),
legend.text.align = 0,
legend.text = element_text(size = 22))+
scale_fill_manual(labels = c("null" = "Null distribution",
"mt_observed"= expression(Observed~italic(Fancm^Delta*""^"2/"*""^Delta*""^"2")),
"wt_observed" = expression(Observed~italic("Fancm"^"+/+" ))),
values = c("null" = "grey",
"mt_observed" = "cornflowerblue",
"wt_observed" = "tan1"))+facet_wrap(.~sampleType)+
theme(strip.background = element_blank(),
strip.text.x = element_blank(),axis.title= element_text(size = 25),
axis.text = element_text(size = 25))+ylab("Density")
p_dbco_dist_scCNV_twopanels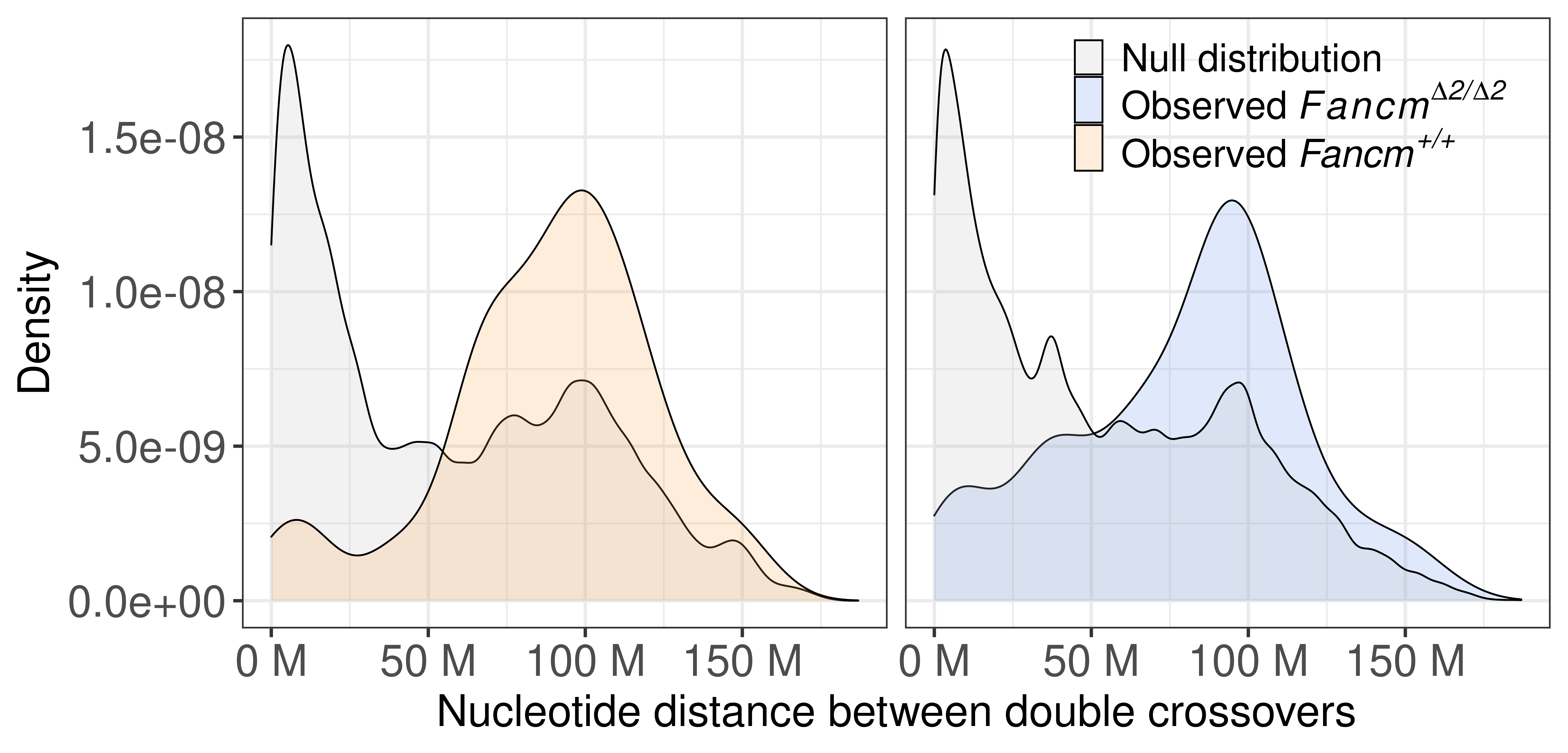
ggsave(filename = "output/outputR/analysisRDS/figures/dbco-dist-scCNV_twopanels.png",
width = 12.5,height = 6,dpi = 300,plot = p_dbco_dist_scCNV_twopanels)BC1F1
Crossover interference
Find chromosomes with double crossovers
bc1f1_samples <- readRDS(file = "output/outputR/analysisRDS/all_rse_count_07-20.rds")chrCOs_bc1f1 <- foreach(chr = paste0("chr",1:19),.combine = "rbind",
.packages = c("BiocGenerics",
"GenomicRanges","SummarizedExperiment")) %dopar% {
tmp <- bc1f1_samples[seqnames(bc1f1_samples)==chr,]
data.frame(barcode = names(colSums(as.matrix(assay(tmp)))),
COs = colSums( as.matrix(assay(tmp))),
Chrom = chr)
}
dbCO_barcodes <- chrCOs_bc1f1[chrCOs_bc1f1$COs==2,] Double crossover distribution
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(bc1f1_samples)[barcode,"sampleGroup"]) %>%
dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE)) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("Male_WT",
"Male_HET",
"Male_KO"),
to =c("Male_non-KO","Male_non-KO","Male_KO"))) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.8, 0.8))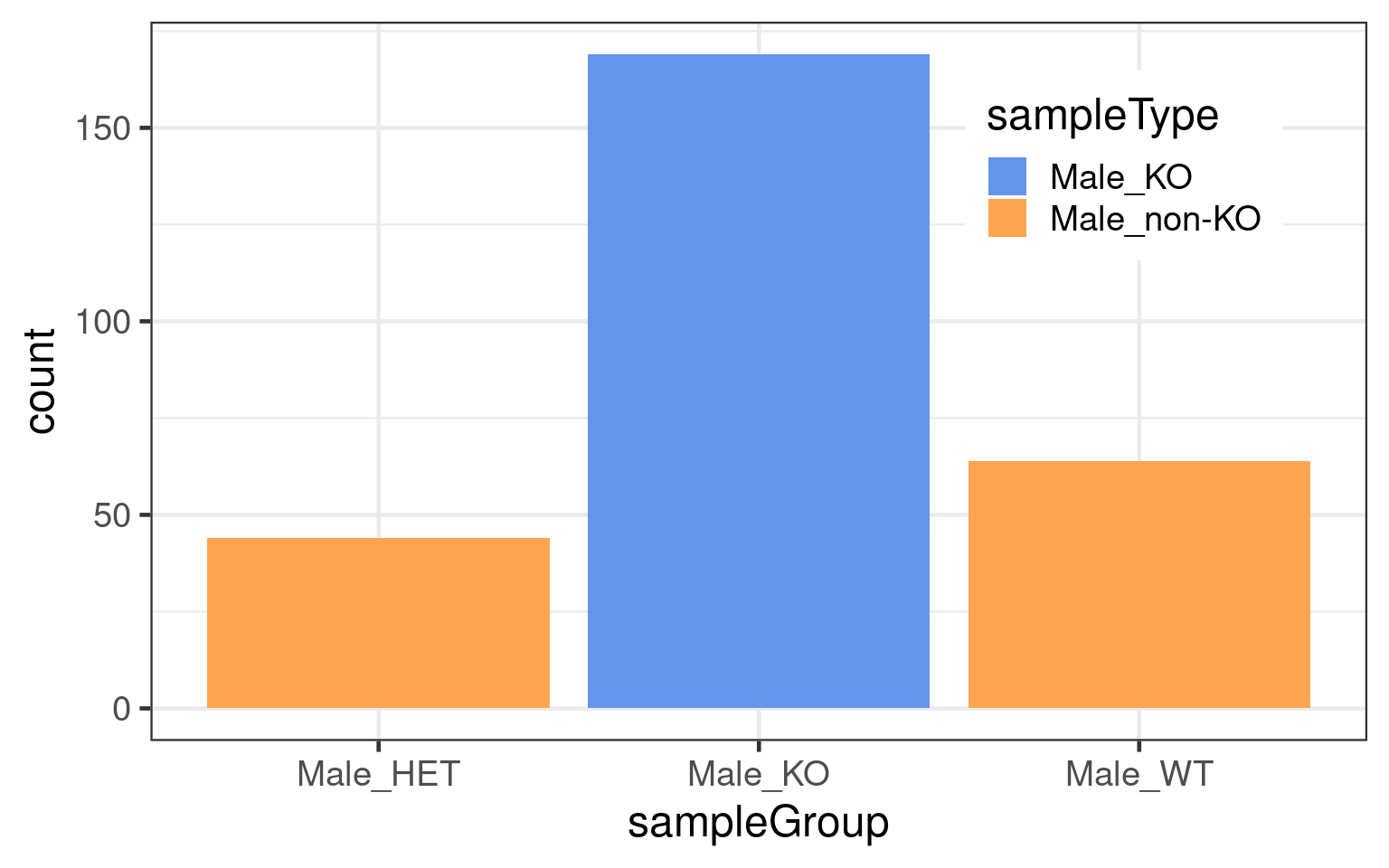
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(bc1f1_samples)[barcode,"sampleGroup"]) %>%
dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE)) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("Male_WT",
"Male_HET",
"Male_KO"),
to =c("Male_non-KO","Male_non-KO","Male_KO"))) %>% ggplot() +
geom_bar(mapping = aes(x = sampleGroup,
fill = sampleType))+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.8, 0.8))+guides(fill =FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.
bc1f1_samples rate of double crossover chromosomes
There might be more sperms from mutant individuals, so we calculate the mean double crossover rate.
nSamples <- data.frame(nSperms = table(bc1f1_samples$sampleGroup))
colnames(nSamples) <- c("sampleGroup","Freq")
nSamples <- nSamples %>% dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE))The number of chromosomes with double crossovers normalised by the number of samples per group
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(bc1f1_samples)[barcode,"sampleGroup"]) %>%
dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE)) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("Male_WT",
"Male_HET",
"Male_KO"),
to =c("Male_non-KO","Male_non-KO","Male_KO"))) %>%
dplyr::group_by(sampleGroup,sampleType) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSamples$sampleGroup,
to = nSamples$Freq)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleType, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.8, 0.8))`summarise()` has grouped output by 'sampleGroup'. You can override using the `.groups` argument.
The following `from` values were not present in `x`: Male_KO, Male_WT
The following `from` values were not present in `x`: Male_HET, Male_WT
The following `from` values were not present in `x`: Male_HET, Male_KO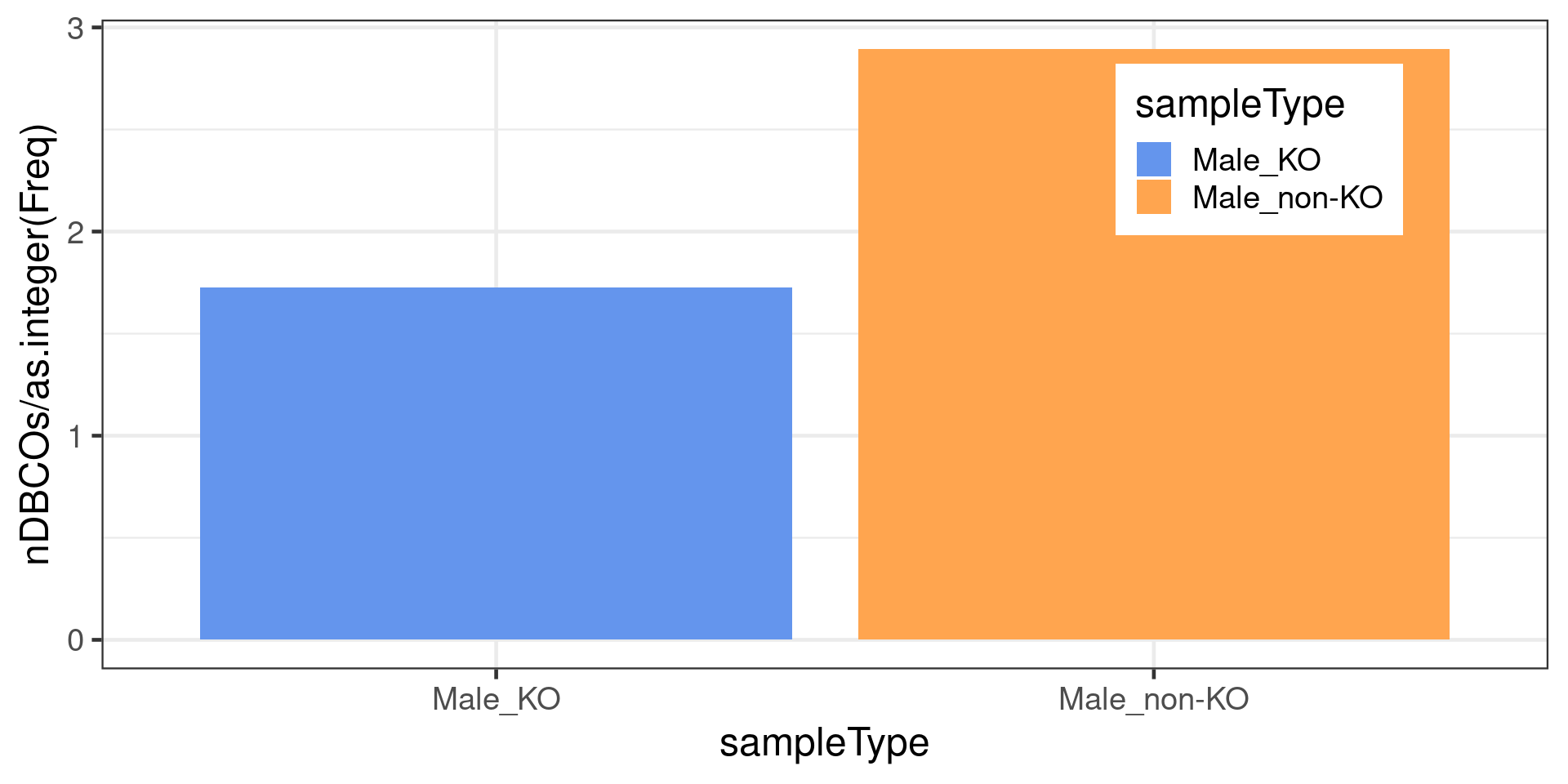
dbCO_barcodes %>% dplyr::mutate(sampleGroup = colData(bc1f1_samples)[barcode,"sampleGroup"]) %>%
dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE)) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("Male_WT",
"Male_HET",
"Male_KO"),
to =c("Male_non-KO","Male_non-KO","Male_KO"))) %>%
dplyr::group_by(sampleGroup,sampleType) %>%
dplyr::summarise(nDBCOs = n()) %>% dplyr::mutate(Freq = plyr::mapvalues(sampleGroup,
from = nSamples$sampleGroup,
to = nSamples$Freq)) %>% ggplot() +
geom_bar(mapping = aes(x = sampleType, y = nDBCOs/as.integer(Freq),
fill = sampleType),
stat = "identity")+theme_bw(base_size = 18)+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.8, 0.8))+guides(fill =FALSE)`summarise()` has grouped output by 'sampleGroup'. You can override using the `.groups` argument.
The following `from` values were not present in `x`: Male_KO, Male_WT
The following `from` values were not present in `x`: Male_HET, Male_WT
The following `from` values were not present in `x`: Male_HET, Male_KOWarning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.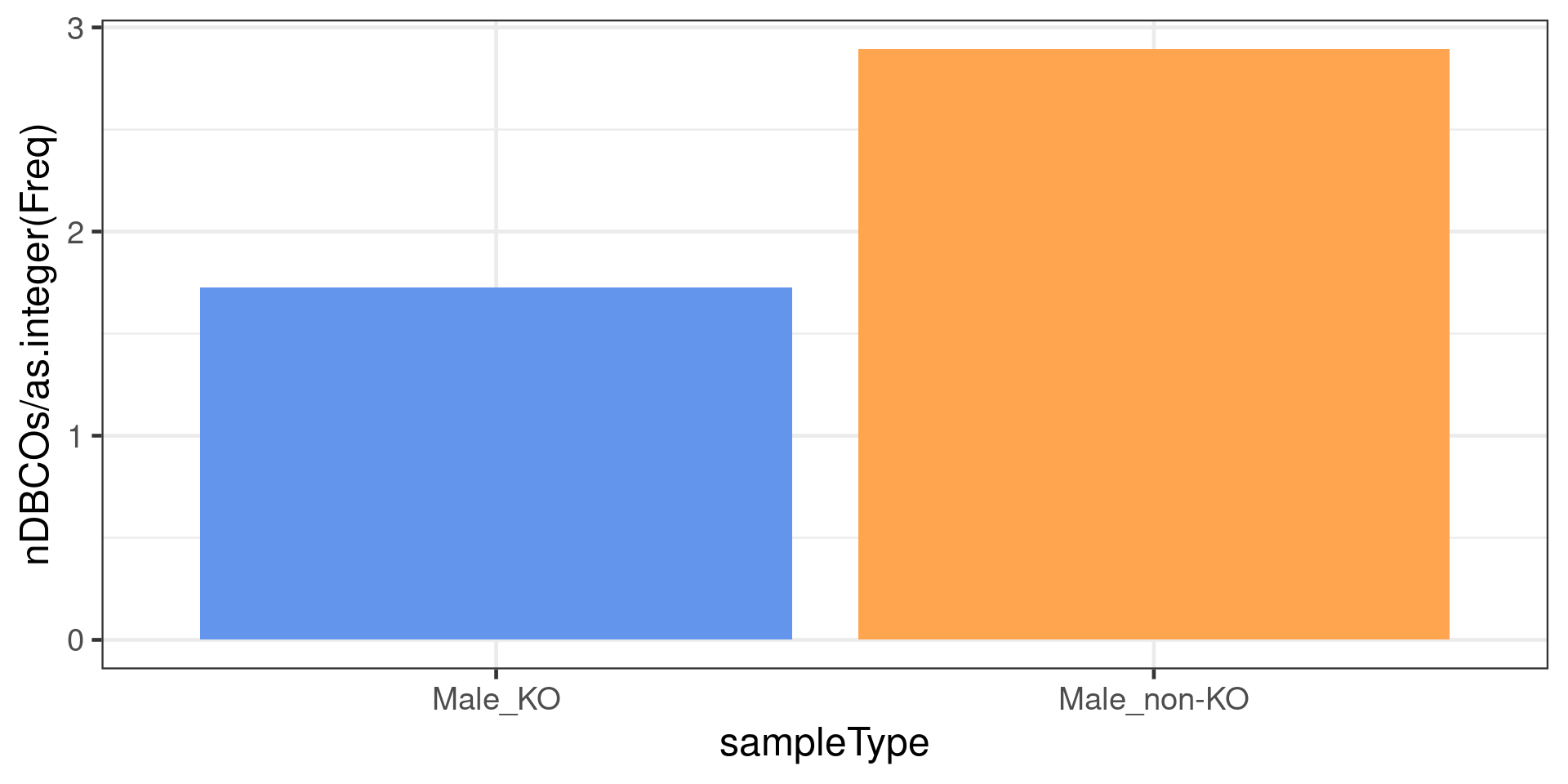
bulk BC1F1 Inter-crossover distances
double_CO_intervals_bc1f1_samples <- foreach(bc = unique(dbCO_barcodes$barcode),.combine = "rbind",
.packages = c("BiocGenerics",
"GenomicRanges","SummarizedExperiment")) %dopar%
{
dd <- data.frame(start_pos =
start(comapr::getCellCORange(bc1f1_samples,
cellBarcode =bc)),
end_pos = end(comapr::getCellCORange(bc1f1_samples,
cellBarcode =bc)),
Chrom = as.character(seqnames(comapr::getCellCORange(bc1f1_samples,
cellBarcode = bc))),
barcode = bc)
keepChrs <- names( table(dd$Chrom)[table(dd$Chrom)==2])
dd[dd$Chrom%in% keepChrs,]
}double_CO_intervals_bc1f1_samples$coID <- paste0(double_CO_intervals_bc1f1_samples$Chrom,"_",
double_CO_intervals_bc1f1_samples$barcode)
double_CO_intervals_bc1f1_samples <- double_CO_intervals_bc1f1_samples %>%
dplyr::mutate(sampleGroup = colData(bc1f1_samples)[barcode,"sampleGroup"]) %>%
dplyr::filter(grepl("Male",sampleGroup,ignore.case = FALSE)) %>%
dplyr::mutate(sampleType = plyr::mapvalues(sampleGroup,
from = c("Male_WT",
"Male_HET",
"Male_KO"),
to =c("Male_non-KO","Male_non-KO","Male_KO")))
secondCO <- double_CO_intervals_bc1f1_samples[duplicated(double_CO_intervals_bc1f1_samples$coID),]
firstCO <- double_CO_intervals_bc1f1_samples[!duplicated(double_CO_intervals_bc1f1_samples$coID),]
double_CO_bc1f1_wide <- merge(firstCO,secondCO,
by = c("coID","sampleGroup","sampleType",
"barcode","Chrom"))double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist))+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples Nucleotide distance between double crossovers ")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.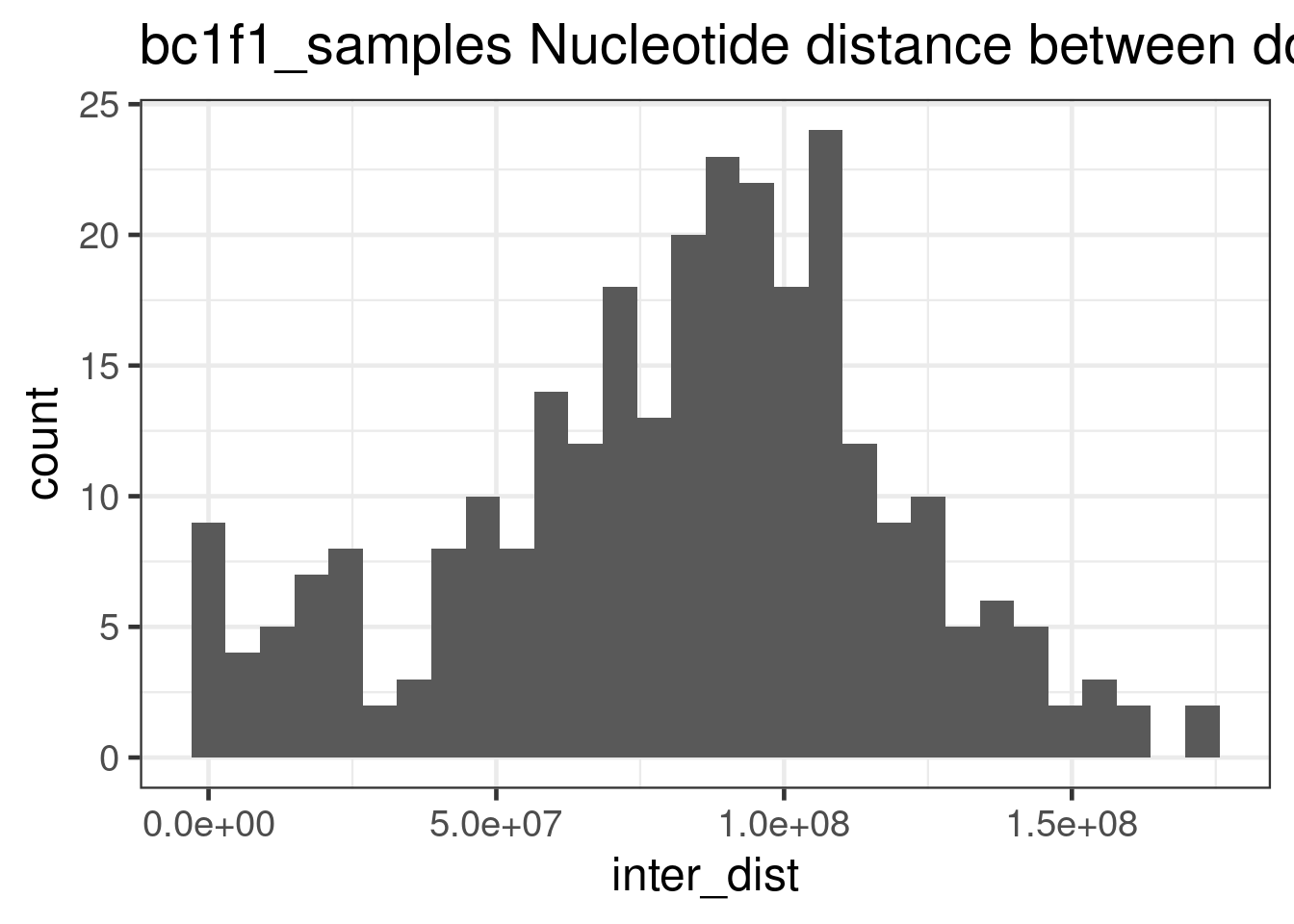
p_double_CO_bc1f1_wide <- double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleType))+theme_bw(base_size = 25) +
scale_x_continuous(labels = scales::unit_format(unit = "M",
scale = 1e-06))+
ggtitle("BC1F1 bulk sequencing pups")+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"),
labels = c("Male_KO"= expression(italic(Fancm^Delta*""^"2/"*""^Delta*""^"2")),
"Male_non-KO" = expression(italic("Fancm"^"+/*" ))))+
theme(legend.position = c(0.78, 0.8),legend.text.align = 0,legend.background = element_blank(),
legend.title = element_blank(),
legend.text = element_text(size = 22),
axis.text =element_text(size = 25))+xlab("Nucleotide distance between double crossovers")+ylab("Count")
p_double_CO_bc1f1_wide`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.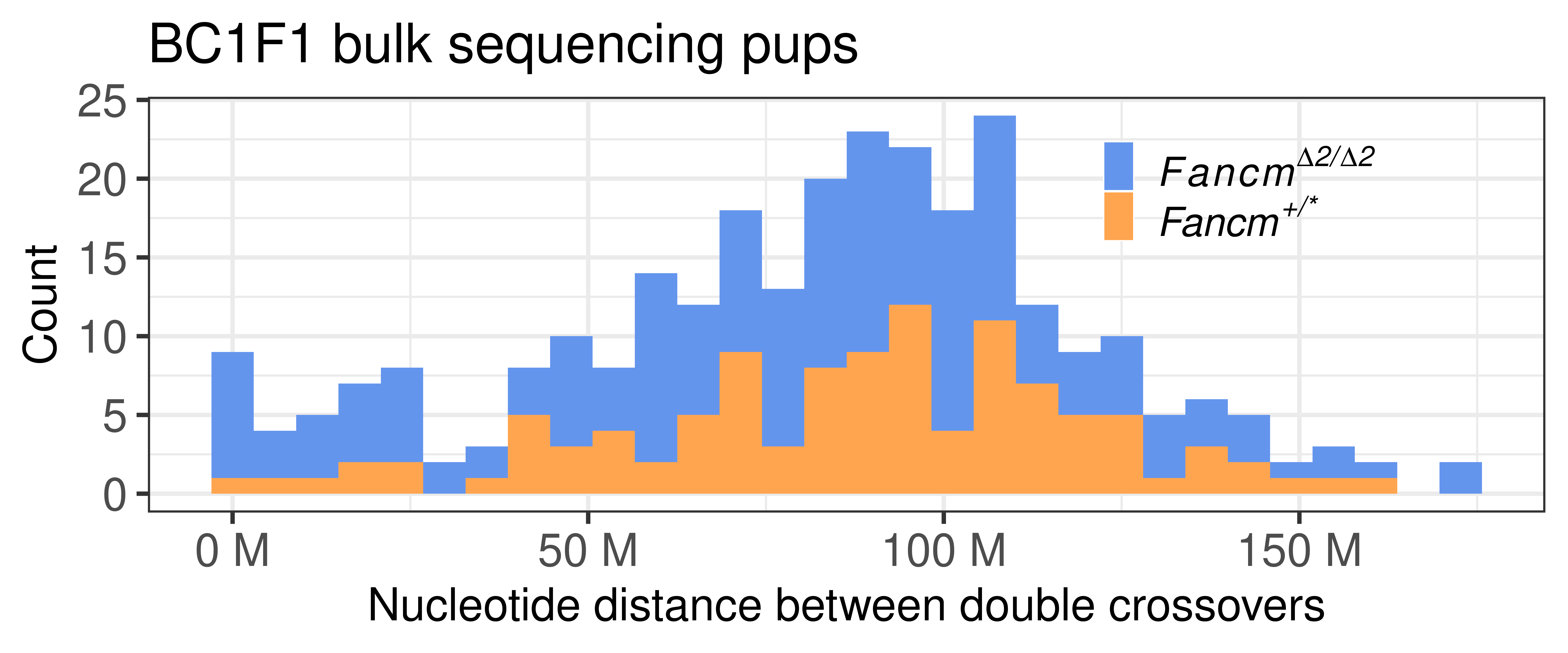
ggsave(filename = "output/outputR/analysisRDS/figures/double_CO_bc1f1_wide-hist.png",
dpi = 300, width = 12,plot = p_double_CO_bc1f1_wide)Number of double crossovers per group
table(double_CO_bc1f1_wide$sampleType)
Male_KO Male_non-KO
175 109 double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_density(mapping = aes(inter_dist,
fill = sampleType),alpha=0.3)+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples Nucleotide distance between double crossovers ")+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.78, 0.8))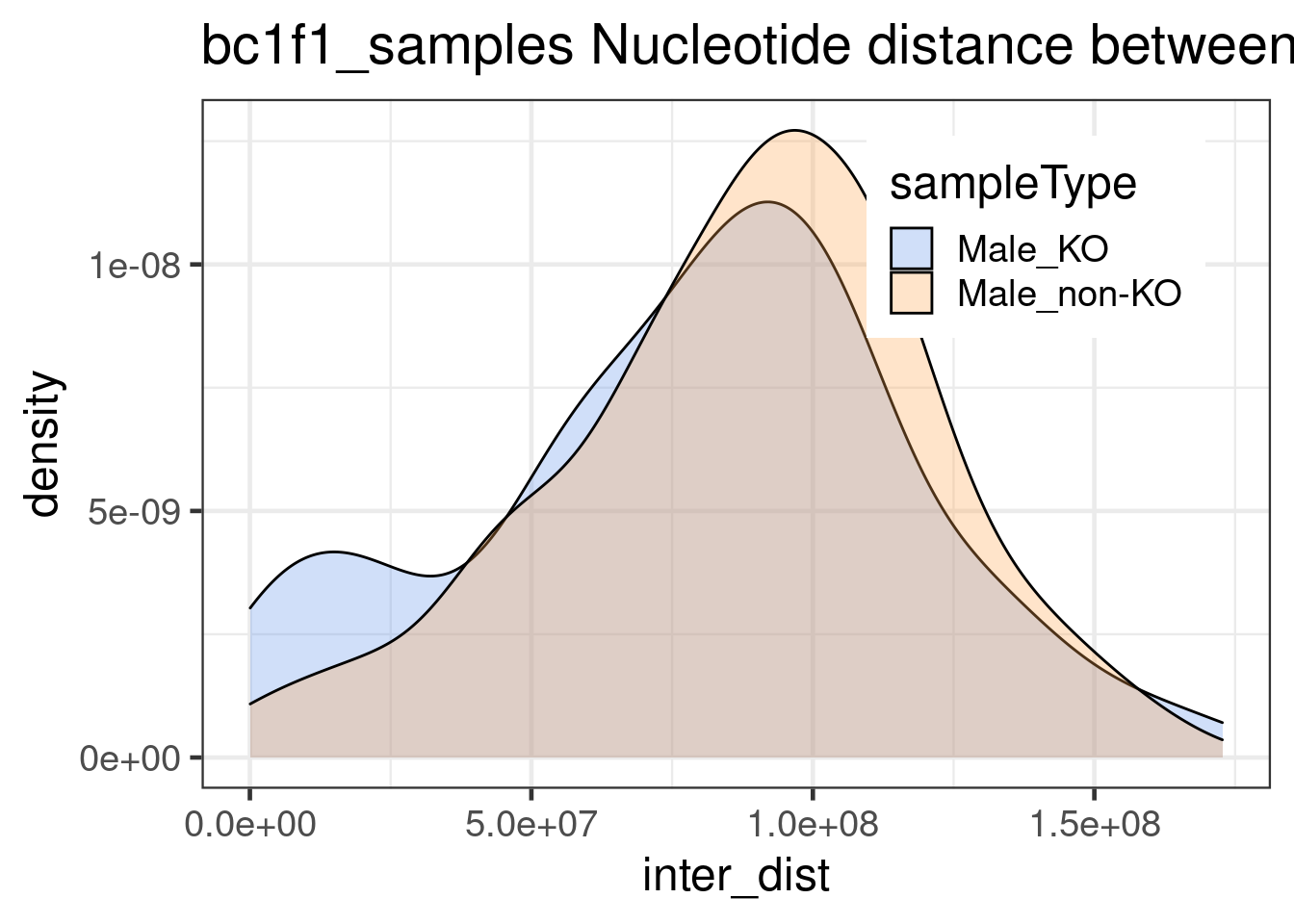
double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_density(mapping = aes(inter_dist,
fill = sampleType),alpha=0.3)+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples Nucleotide distance between double crossovers ")+
scale_fill_manual(values = c("Male_KO"="cornflowerblue",
"Male_non-KO" = "tan1"))+
theme(legend.position = c(0.78, 0.8))+guides(fill =FALSE)Warning: `guides(<scale> = FALSE)` is deprecated. Please use `guides(<scale> =
"none")` instead.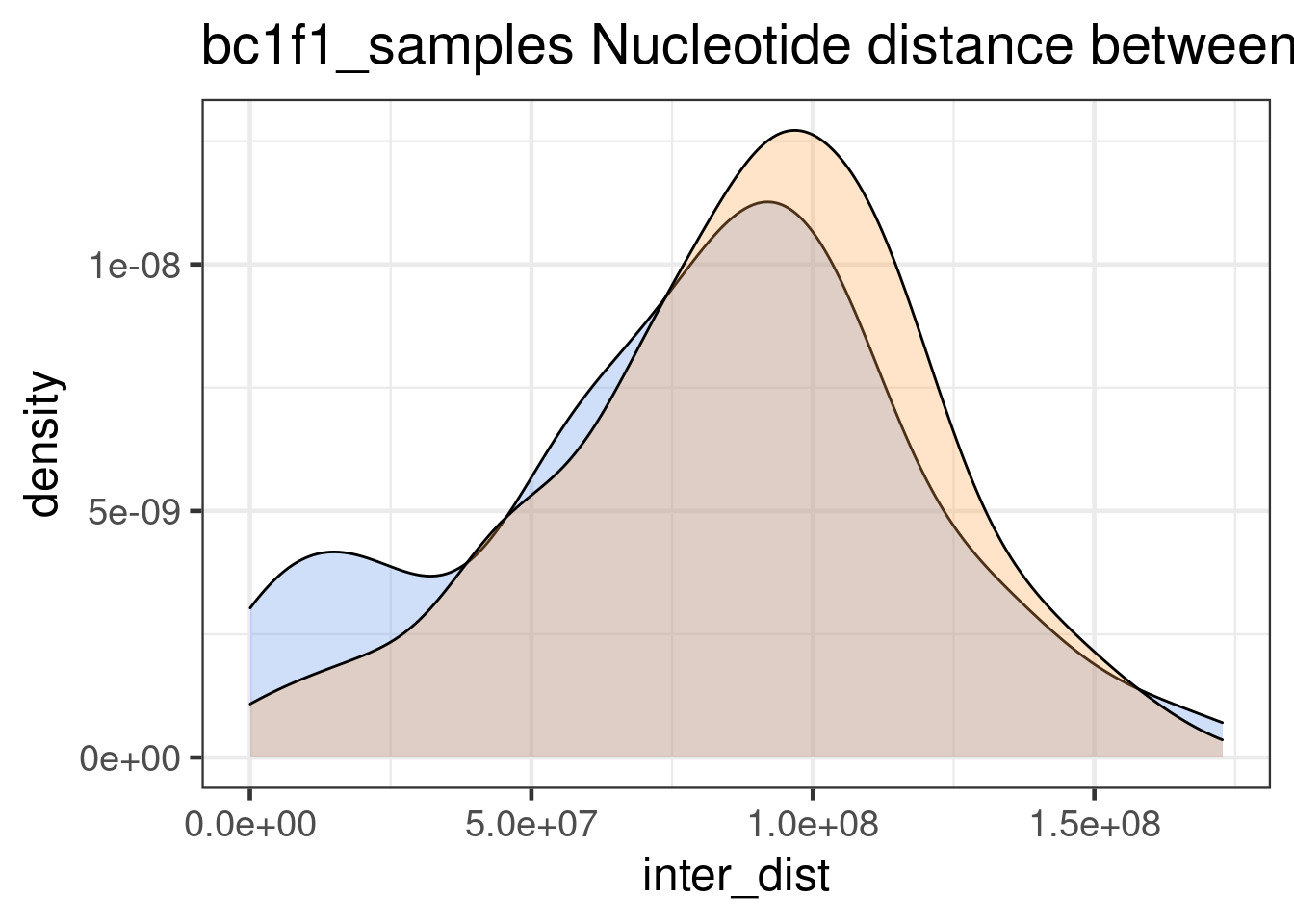
Interdistances from mutant double crossovers are smaller than Inter-distances from wildtype double crossovers. (by Wilcox.test)
mt_dist <- double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter( sampleType == "Male_KO") %>% dplyr::select(inter_dist)
wt_dist <- double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter( sampleType == "Male_non-KO") %>% dplyr::select(inter_dist)
wilcox.test(mt_dist$inter_dist,wt_dist$inter_dist,alternative = "less")
Wilcoxon rank sum test with continuity correction
data: mt_dist$inter_dist and wt_dist$inter_dist
W = 8187, p-value = 0.02244
alternative hypothesis: true location shift is less than 0double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes(inter_dist,
fill = sampleGroup))+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples Nucleotide distance between double crossovers ")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.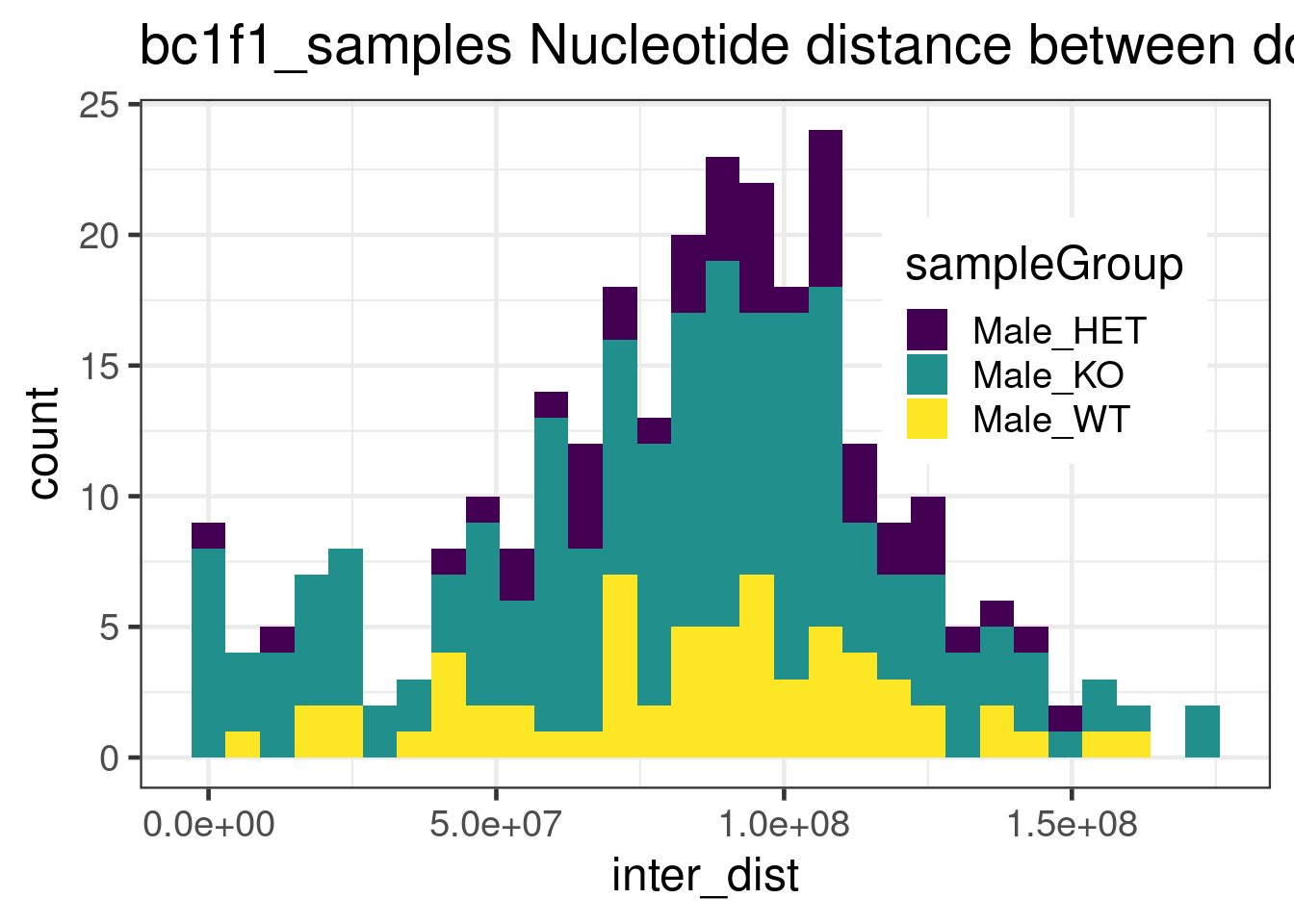
crossover interference by permuting the crossover positions across the chromosomes
mutant bc1f1
Permute 1000 times for mutant/wildtypes
B <- 1000
set.seed(2021)
mutant_bulk_permute <- foreach(b = seq(B), .combine = "rbind") %dopar% {
pm <- lapply(unique(firstCO[firstCO$sampleType=="Male_KO","Chrom"]),
function(chr){
permuteOnce(firstCOs = firstCO[firstCO$sampleType=="Male_KO",],
secondCOs = secondCO[secondCO$sampleType=="Male_KO",],
chr = chr)
})
pm <- do.call(rbind,pm)
}
#head(mutant_permute)wt_bulk_permute <- foreach(b = seq(B), .combine = "rbind") %dopar% {
pm <- lapply(unique(firstCO[firstCO$sampleType=="Male_non-KO","Chrom"]),
function(chr){
permuteOnce(firstCOs = firstCO[firstCO$sampleType=="Male_non-KO",],
secondCOs = secondCO[secondCO$sampleType=="Male_non-KO",],
chr = chr)
})
pm <- do.call(rbind,pm)
}Plot Null distribution of inter-crossover distance versus observed inter-crossover distances
wt_bulk_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes( abs(inter_dist)))+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples Inter-distances of double \n crossovers (null) wildtype ")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()+xlab("inter crossover distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.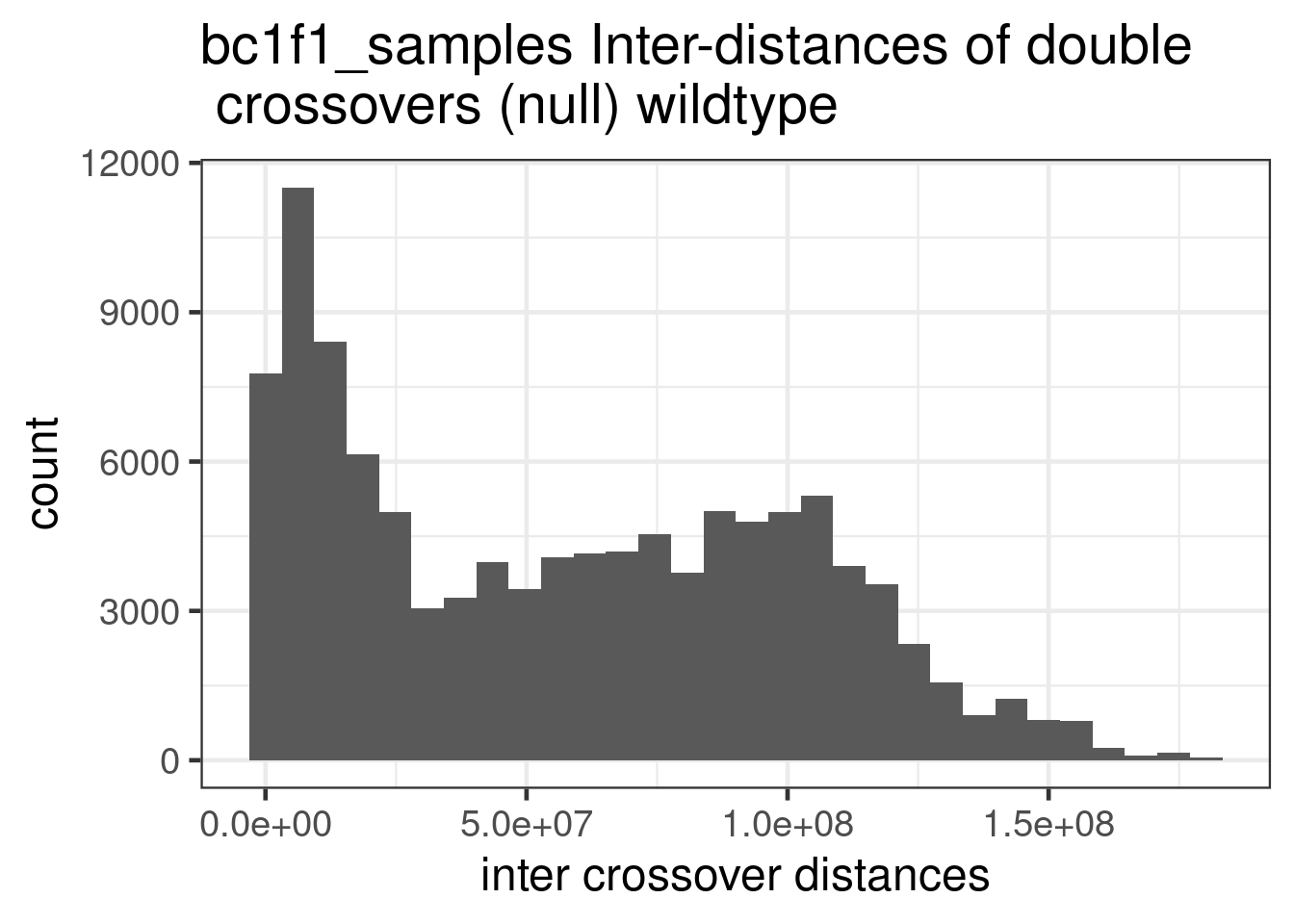
mutant_bulk_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
ggplot() +
geom_histogram(mapping = aes( abs(inter_dist)))+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples inter distances of double \n crossovers (null) mutant ")+
theme(legend.position = c(0.8, 0.65))+scale_fill_viridis_d()+xlab("inter crossover distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.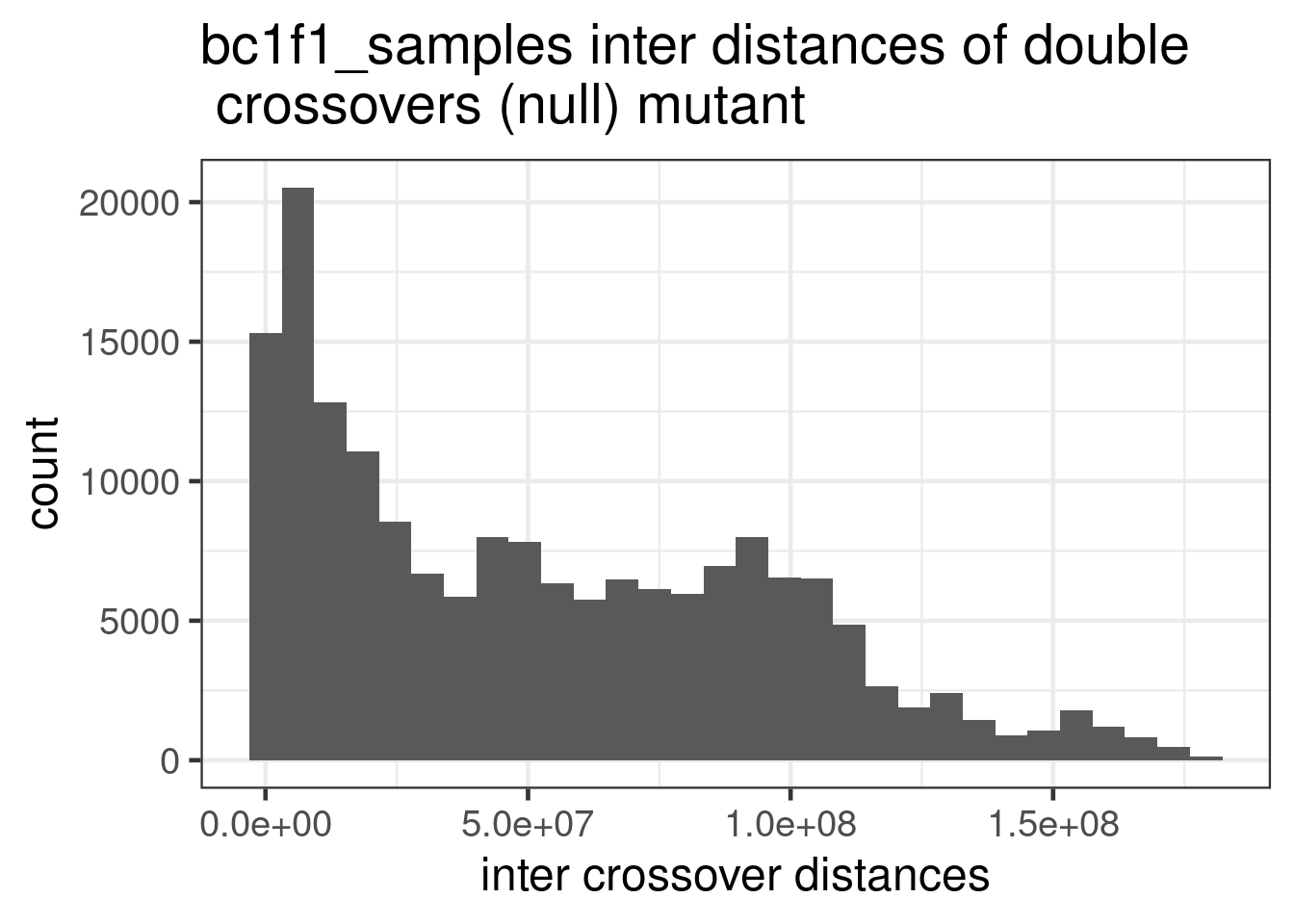
Comparing the observed versus the null for mutant
mutant_bulk_permute <- mutant_bulk_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>% dplyr::mutate(source = "null")
mutant_double_CO_bc1f1_wide <- double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter(sampleType== "Male_KO") %>%
dplyr::mutate(source = "observed") %>%
dplyr::select(colnames(mutant_bulk_permute))p_mt_null_observed_bulk <- rbind(mutant_bulk_permute, mutant_double_CO_bc1f1_wide) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.3)+theme_bw(base_size = 18)+
ggtitle("BC1F1 bulk sequencing pups")+xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.7, 0.86),
legend.title = element_blank(),
legend.background = element_blank())+
scale_fill_manual(labels = c("null" = "Null distribution",
"observed" = "observed (Fancm-/-)"),
values = c("null" = "grey",
"observed" = "cornflowerblue"))
p_mt_null_observed_bulk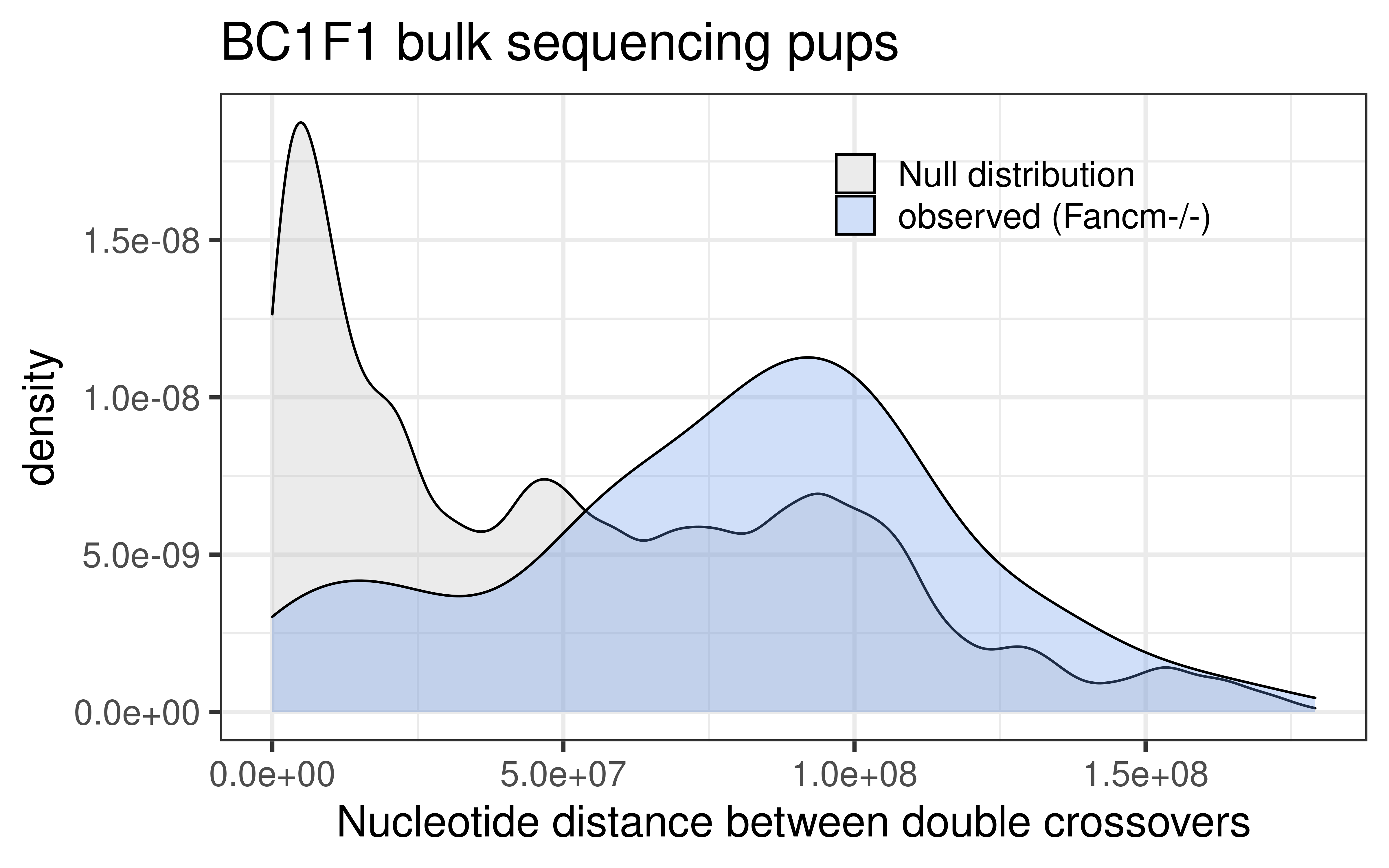
Comparing the observed versus the null for wildtype
wt_bulk_permute <- wt_bulk_permute %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>% dplyr::mutate(source = "null")
wt_double_CO_bc1f1_wide <- double_CO_bc1f1_wide %>% dplyr::mutate(firstMid = 0.5*(end_pos.x + start_pos.x),
secondMid = 0.5*(end_pos.y + start_pos.y),
inter_dist = secondMid - firstMid) %>%
dplyr::filter(sampleType== "Male_non-KO") %>%
dplyr::mutate(source = "observed") %>%
dplyr::select(colnames(wt_bulk_permute))p_wt_null_observed_bulk <- rbind(wt_bulk_permute, wt_double_CO_bc1f1_wide) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.2)+theme_bw(base_size = 18) +
ggtitle("bc1f1_samples inter distances of double crossovers \n (null versus observed) wildtype")+
theme(legend.position = c(0.8, 0.65))+
xlab("inter crossover distances")+
ggtitle("BC1F1 bulk sequencing pups")+xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.7, 0.89),
legend.title = element_blank(),
legend.background = element_blank())+
scale_fill_manual(labels = c("null" = "Null distribution",
"observed" = "observed (Fancm+/*)"),
values = c("null" = "grey",
"observed" = "tan1"))
p_wt_null_observed_bulk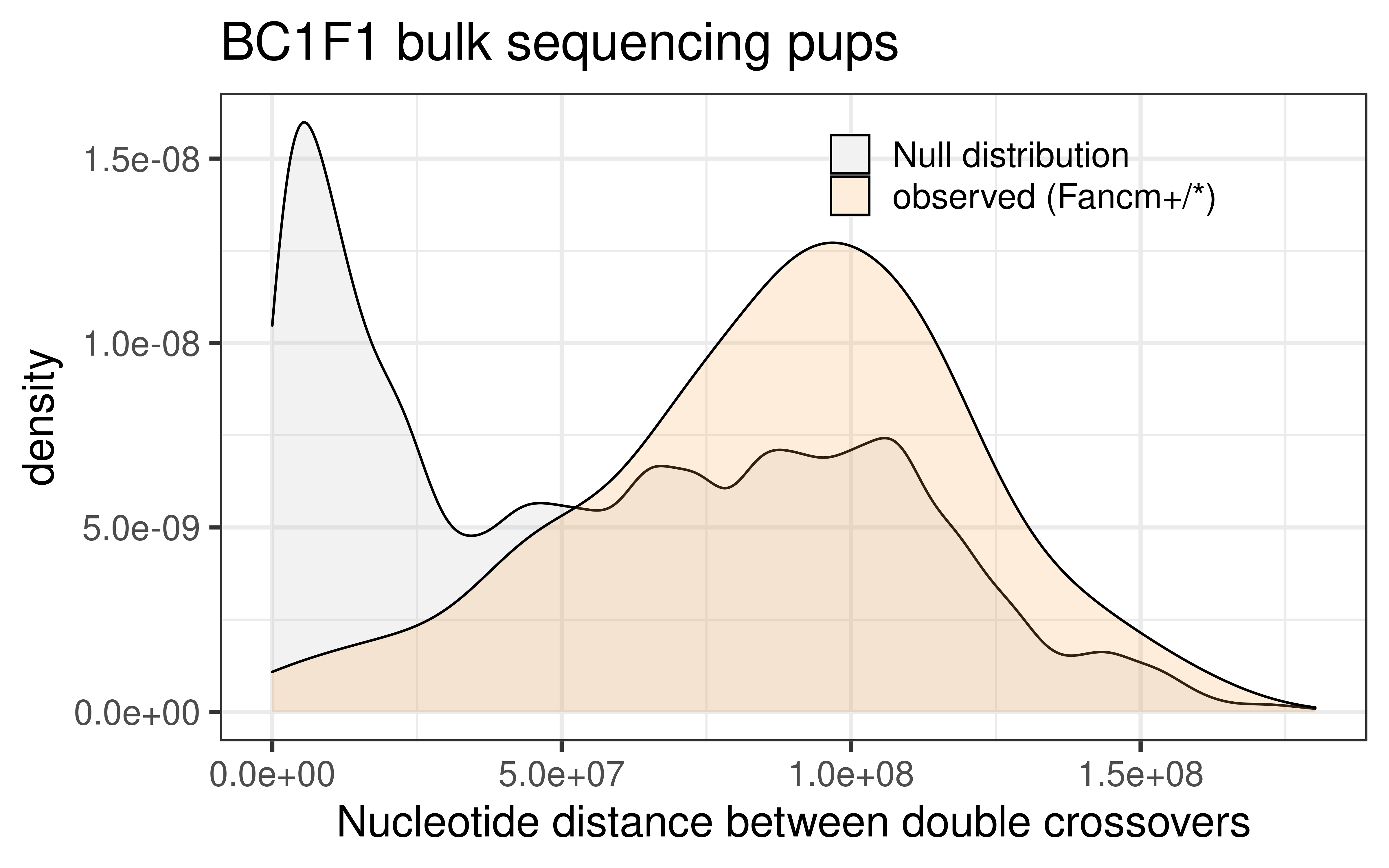
wt_bulk_panel <- rbind(wt_bulk_permute, wt_double_CO_bc1f1_wide)
wt_bulk_panel$source <- plyr::mapvalues(wt_bulk_panel$source,from =c("null","observed"),
to = c("null","wt_observed"))
wt_bulk_panel$sampleType <- "wt"
mt_bulk_panel <- rbind(mutant_bulk_permute, mutant_double_CO_bc1f1_wide)
mt_bulk_panel$source <- plyr::mapvalues(mt_bulk_panel$source,from =c("null","observed"),
to = c("null","mt_observed"))
mt_bulk_panel$sampleType <- "mt"
p_dbco_dist_bc1f1_twopanels <- rbind(mt_bulk_panel,wt_bulk_panel) %>% mutate(sampleType = factor(sampleType,levels = c("wt","mt"))) %>%
ggplot() +
geom_density(mapping = aes(abs(inter_dist),
fill = source),alpha = 0.2)+
scale_x_continuous(labels = scales::unit_format(unit = "M",
scale = 1e-06))+
theme_bw(base_size = 20)+
theme_bw(base_size = 20) +
xlab("Nucleotide distance between double crossovers")+
theme(legend.position = c(0.8, 0.87),
legend.title = element_blank(),
legend.background = element_blank(),
legend.text.align = 0,
legend.text = element_text(size = 22))+
scale_fill_manual(labels = c("null" = "Null distribution",
"mt_observed"= expression(Observed~italic(Fancm^Delta*""^"2/"*""^Delta*""^"2")),
"wt_observed" = expression(Observed~italic("Fancm"^"+/*" ))),
values = c("null" = "grey",
"mt_observed" = "cornflowerblue",
"wt_observed" = "tan1"))+facet_wrap(.~sampleType)+
theme(strip.background = element_blank(),
strip.text.x = element_blank(),
axis.title= element_text(size = 25),
axis.text = element_text(size = 25))+ylab("Density")
p_dbco_dist_bc1f1_twopanels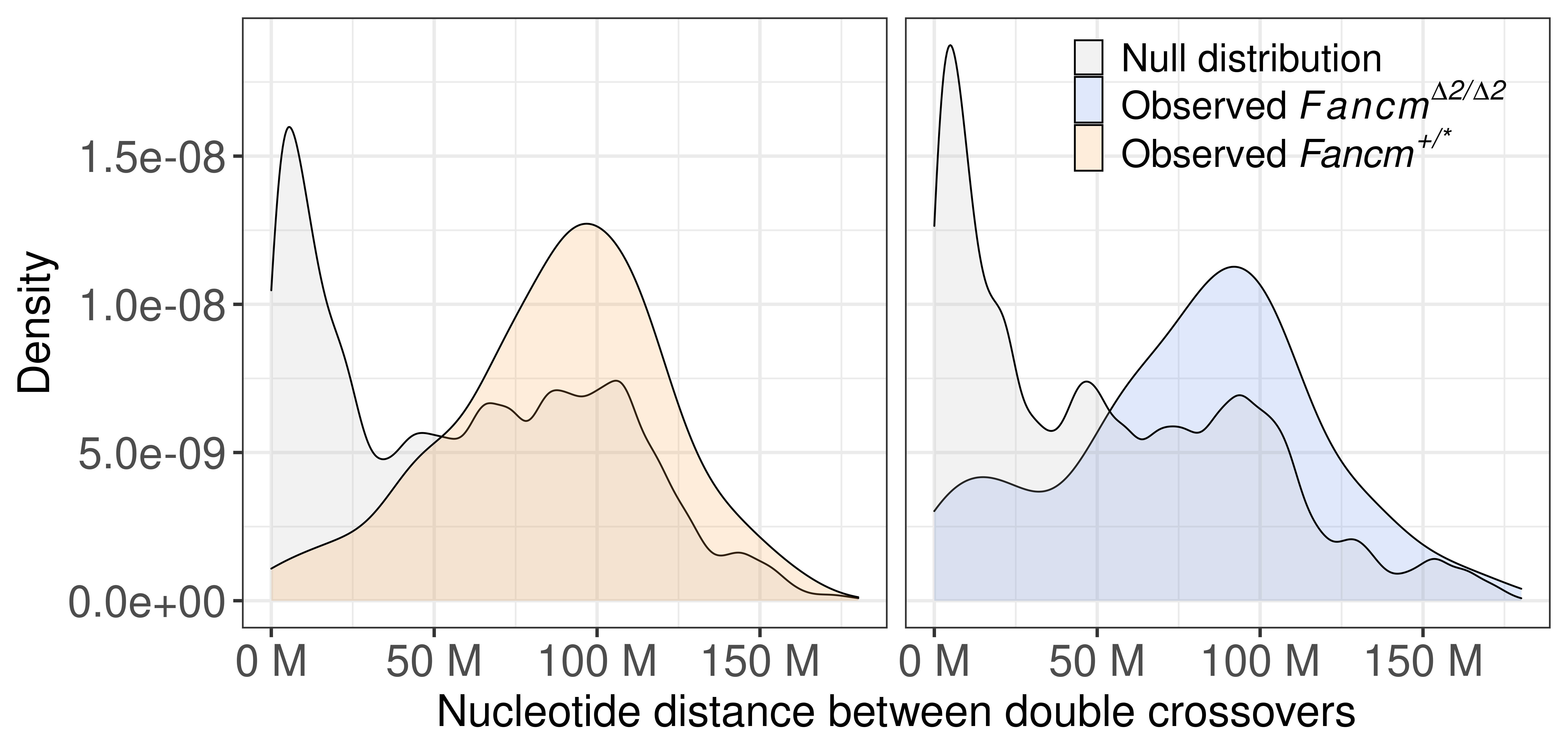
ggsave(filename = "output/outputR/analysisRDS/figures/dbco_dist_bc1f1_twopanels.png",
width = 12.5,height = 6,dpi = 300,plot = p_dbco_dist_bc1f1_twopanels)four plots
arg_mchrs1 <- arrangeGrob(p_wt_null_observed,p_mt_null_observed,
p_wt_null_observed_bulk,p_mt_null_observed_bulk)
arg_mchrs_sccnv <- arrangeGrob(p_wt_null_observed,p_mt_null_observed)
arg_mchrs_bulk <- arrangeGrob(p_wt_null_observed_bulk,p_mt_null_observed_bulk)
grid.arrange(arg_mchrs_sccnv)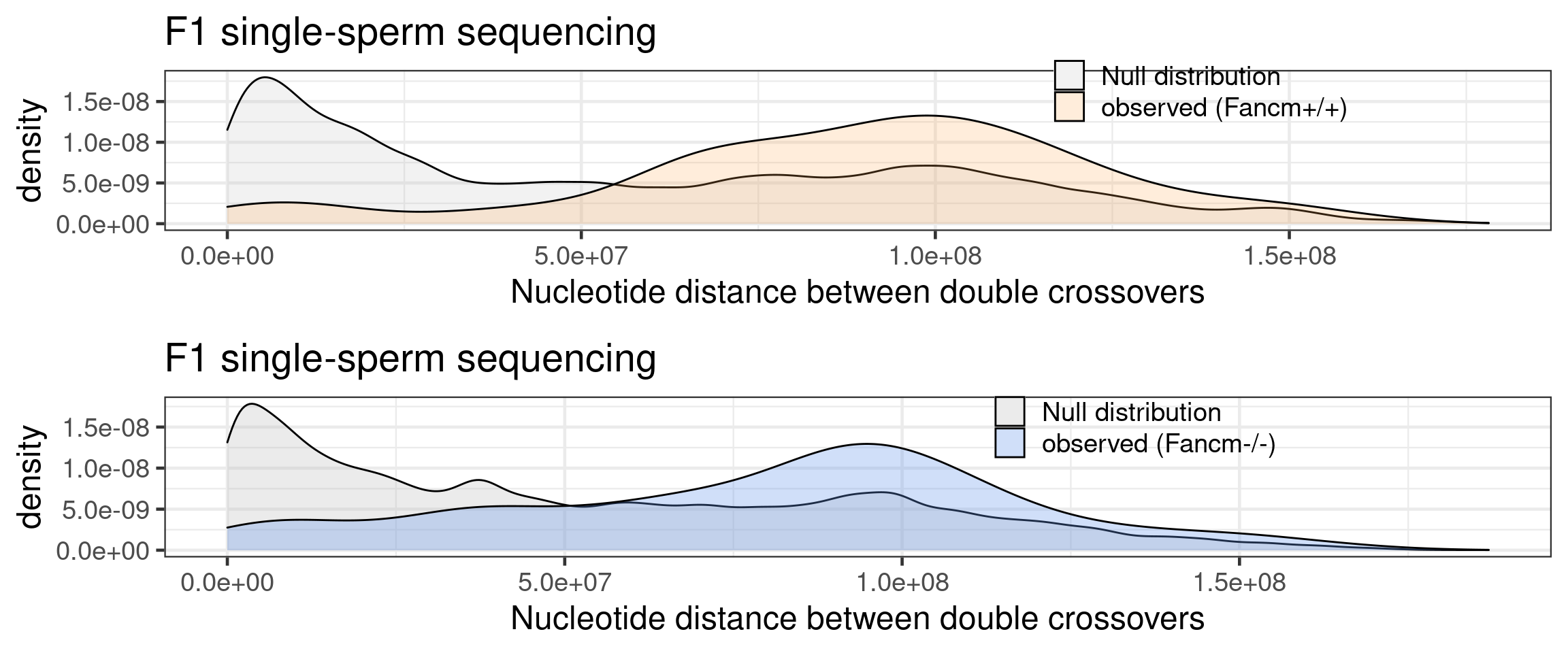
grid.arrange(arg_mchrs_bulk)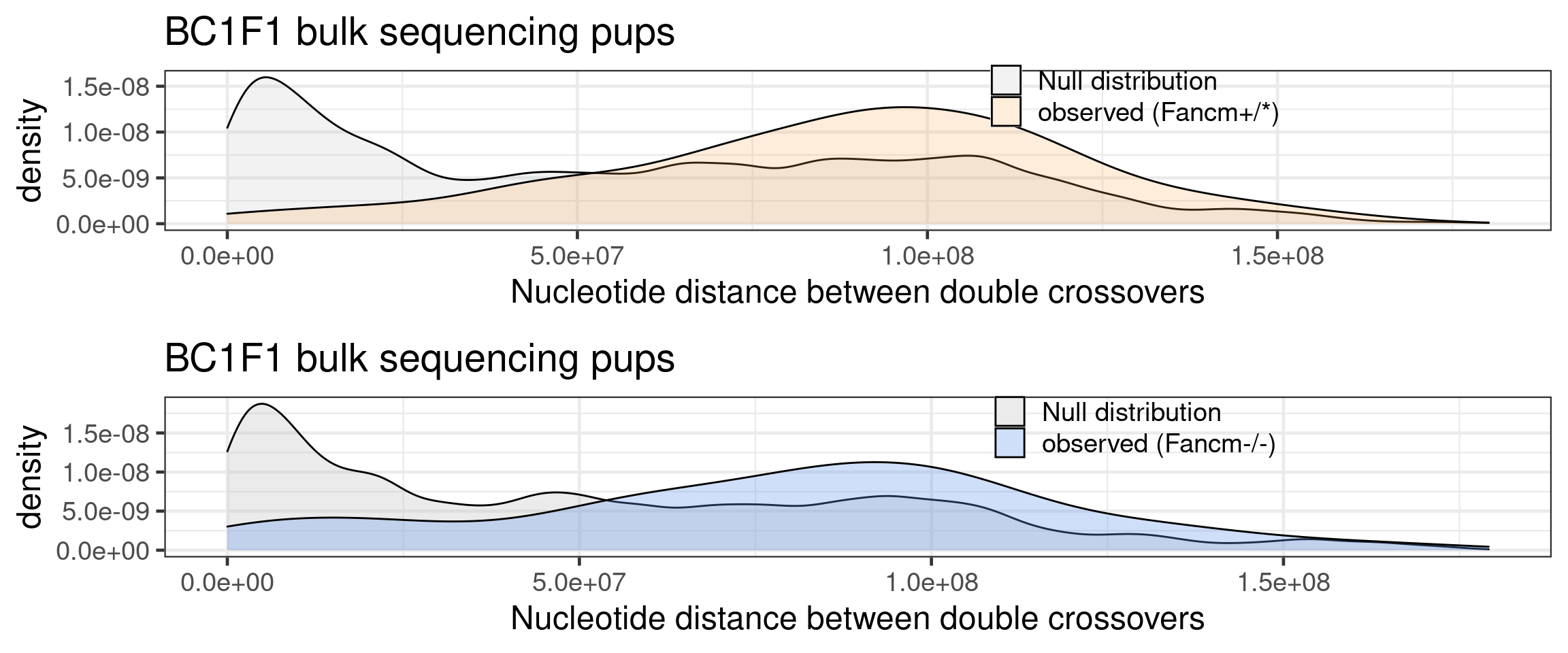
Session info
sessionInfo()R version 4.1.2 (2021-11-01)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Rocky Linux 8.5 (Green Obsidian)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel grid stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] doParallel_1.0.17 iterators_1.0.14
[3] foreach_1.5.2 Gviz_1.38.3
[5] gridExtra_2.3 SummarizedExperiment_1.24.0
[7] Biobase_2.54.0 GenomicRanges_1.46.1
[9] GenomeInfoDb_1.30.1 IRanges_2.28.0
[11] S4Vectors_0.32.3 BiocGenerics_0.40.0
[13] MatrixGenerics_1.6.0 matrixStats_0.61.0
[15] dplyr_1.0.7 comapr_0.99.43
[17] ggrepel_0.9.1 ggplot2_3.3.5
[19] readxl_1.3.1
loaded via a namespace (and not attached):
[1] backports_1.4.1 circlize_0.4.13 Hmisc_4.6-0
[4] workflowr_1.7.0 BiocFileCache_2.2.1 plyr_1.8.6
[7] lazyeval_0.2.2 splines_4.1.2 BiocParallel_1.28.3
[10] digest_0.6.29 ensembldb_2.18.3 htmltools_0.5.2
[13] fansi_1.0.2 magrittr_2.0.2 checkmate_2.0.0
[16] memoise_2.0.1 BSgenome_1.62.0 cluster_2.1.2
[19] Biostrings_2.62.0 prettyunits_1.1.1 jpeg_0.1-9
[22] colorspace_2.0-2 blob_1.2.2 rappdirs_0.3.3
[25] xfun_0.29 crayon_1.4.2 RCurl_1.98-1.5
[28] jsonlite_1.7.3 survival_3.2-13 VariantAnnotation_1.40.0
[31] glue_1.6.1 gtable_0.3.0 zlibbioc_1.40.0
[34] XVector_0.34.0 DelayedArray_0.20.0 shape_1.4.6
[37] scales_1.1.1 DBI_1.1.2 Rcpp_1.0.8
[40] viridisLite_0.4.0 progress_1.2.2 htmlTable_2.4.0
[43] foreign_0.8-81 bit_4.0.4 Formula_1.2-4
[46] htmlwidgets_1.5.4 httr_1.4.2 RColorBrewer_1.1-2
[49] ellipsis_0.3.2 farver_2.1.0 pkgconfig_2.0.3
[52] XML_3.99-0.8 nnet_7.3-16 dbplyr_2.1.1
[55] utf8_1.2.2 labeling_0.4.2 tidyselect_1.1.1
[58] rlang_1.0.0 reshape2_1.4.4 later_1.3.0
[61] AnnotationDbi_1.56.2 munsell_0.5.0 cellranger_1.1.0
[64] tools_4.1.2 cachem_1.0.6 cli_3.1.1
[67] generics_0.1.1 RSQLite_2.2.9 evaluate_0.14
[70] stringr_1.4.0 fastmap_1.1.0 yaml_2.2.2
[73] knitr_1.37 bit64_4.0.5 fs_1.5.2
[76] purrr_0.3.4 KEGGREST_1.34.0 AnnotationFilter_1.18.0
[79] xml2_1.3.3 biomaRt_2.50.3 compiler_4.1.2
[82] rstudioapi_0.13 plotly_4.10.0 filelock_1.0.2
[85] curl_4.3.2 png_0.1-7 tibble_3.1.6
[88] stringi_1.7.6 highr_0.9 GenomicFeatures_1.46.4
[91] lattice_0.20-45 ProtGenerics_1.26.0 Matrix_1.4-0
[94] vctrs_0.3.8 pillar_1.6.5 lifecycle_1.0.1
[97] jquerylib_0.1.4 GlobalOptions_0.1.2 data.table_1.14.2
[100] bitops_1.0-7 httpuv_1.6.5 rtracklayer_1.54.0
[103] R6_2.5.1 BiocIO_1.4.0 latticeExtra_0.6-29
[106] promises_1.2.0.1 codetools_0.2-18 dichromat_2.0-0
[109] assertthat_0.2.1 rprojroot_2.0.2 rjson_0.2.21
[112] withr_2.4.3 GenomicAlignments_1.30.0 Rsamtools_2.10.0
[115] GenomeInfoDbData_1.2.7 hms_1.1.1 rpart_4.1-15
[118] tidyr_1.2.0 rmarkdown_2.11 git2r_0.29.0
[121] biovizBase_1.42.0 base64enc_0.1-3 restfulr_0.0.13
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.1.2 (2021-11-01)
os Rocky Linux 8.5 (Green Obsidian)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2022-03-22
pandoc 2.11.4 @ /usr/lib/rstudio-server/bin/pandoc/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
AnnotationDbi 1.56.2 2021-11-09 [1] Bioconductor
AnnotationFilter 1.18.0 2021-10-26 [1] Bioconductor
assertthat 0.2.1 2019-03-21 [1] CRAN (R 4.1.2)
backports 1.4.1 2021-12-13 [1] CRAN (R 4.1.2)
base64enc 0.1-3 2015-07-28 [1] CRAN (R 4.1.2)
Biobase * 2.54.0 2021-10-26 [1] Bioconductor
BiocFileCache 2.2.1 2022-01-23 [1] Bioconductor
BiocGenerics * 0.40.0 2021-10-26 [1] Bioconductor
BiocIO 1.4.0 2021-10-26 [1] Bioconductor
BiocParallel 1.28.3 2021-12-09 [1] Bioconductor
biomaRt 2.50.3 2022-02-03 [1] Bioconductor
Biostrings 2.62.0 2021-10-26 [1] Bioconductor
biovizBase 1.42.0 2021-10-26 [1] Bioconductor
bit 4.0.4 2020-08-04 [1] CRAN (R 4.1.2)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.1.2)
bitops 1.0-7 2021-04-24 [1] CRAN (R 4.1.2)
blob 1.2.2 2021-07-23 [1] CRAN (R 4.1.2)
brio 1.1.3 2021-11-30 [1] CRAN (R 4.1.0)
BSgenome 1.62.0 2021-10-26 [1] Bioconductor
cachem 1.0.6 2021-08-19 [1] CRAN (R 4.1.0)
callr 3.7.0 2021-04-20 [1] CRAN (R 4.1.2)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.1.2)
checkmate 2.0.0 2020-02-06 [1] CRAN (R 4.1.0)
circlize 0.4.13 2021-06-09 [1] CRAN (R 4.1.0)
cli 3.1.1 2022-01-20 [1] CRAN (R 4.1.2)
cluster 2.1.2 2021-04-17 [2] CRAN (R 4.1.2)
codetools 0.2-18 2020-11-04 [2] CRAN (R 4.1.2)
colorspace 2.0-2 2021-06-24 [1] CRAN (R 4.1.2)
comapr * 0.99.43 2022-03-09 [1] Github (ruqianl/comapr@915d97c)
crayon 1.4.2 2021-10-29 [1] CRAN (R 4.1.2)
curl 4.3.2 2021-06-23 [1] CRAN (R 4.1.2)
data.table 1.14.2 2021-09-27 [1] CRAN (R 4.1.2)
DBI 1.1.2 2021-12-20 [1] CRAN (R 4.1.2)
dbplyr 2.1.1 2021-04-06 [1] CRAN (R 4.1.2)
DelayedArray 0.20.0 2021-10-26 [1] Bioconductor
desc 1.4.0 2021-09-28 [1] CRAN (R 4.1.0)
devtools 2.4.3 2021-11-30 [1] CRAN (R 4.1.0)
dichromat 2.0-0 2013-01-24 [1] CRAN (R 4.1.0)
digest 0.6.29 2021-12-01 [1] CRAN (R 4.1.2)
doParallel * 1.0.17 2022-02-07 [1] CRAN (R 4.1.0)
dplyr * 1.0.7 2021-06-18 [1] CRAN (R 4.1.2)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.1.2)
ensembldb 2.18.3 2022-01-13 [1] Bioconductor
evaluate 0.14 2019-05-28 [1] CRAN (R 4.1.2)
fansi 1.0.2 2022-01-14 [1] CRAN (R 4.1.2)
farver 2.1.0 2021-02-28 [1] CRAN (R 4.1.2)
fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.1.2)
filelock 1.0.2 2018-10-05 [1] CRAN (R 4.1.0)
foreach * 1.5.2 2022-02-02 [1] CRAN (R 4.1.0)
foreign 0.8-81 2020-12-22 [2] CRAN (R 4.1.2)
Formula 1.2-4 2020-10-16 [1] CRAN (R 4.1.0)
fs 1.5.2 2021-12-08 [1] CRAN (R 4.1.2)
generics 0.1.1 2021-10-25 [1] CRAN (R 4.1.2)
GenomeInfoDb * 1.30.1 2022-01-30 [1] Bioconductor
GenomeInfoDbData 1.2.7 2022-01-28 [1] Bioconductor
GenomicAlignments 1.30.0 2021-10-26 [1] Bioconductor
GenomicFeatures 1.46.4 2022-01-20 [1] Bioconductor
GenomicRanges * 1.46.1 2021-11-18 [1] Bioconductor
ggplot2 * 3.3.5 2021-06-25 [1] CRAN (R 4.1.2)
ggrepel * 0.9.1 2021-01-15 [1] CRAN (R 4.1.2)
git2r 0.29.0 2021-11-22 [1] CRAN (R 4.1.2)
GlobalOptions 0.1.2 2020-06-10 [1] CRAN (R 4.1.0)
glue 1.6.1 2022-01-22 [1] CRAN (R 4.1.2)
gridExtra * 2.3 2017-09-09 [1] CRAN (R 4.1.0)
gtable 0.3.0 2019-03-25 [1] CRAN (R 4.1.2)
Gviz * 1.38.3 2022-01-23 [1] Bioconductor
highr 0.9 2021-04-16 [1] CRAN (R 4.1.2)
Hmisc 4.6-0 2021-10-07 [1] CRAN (R 4.1.0)
hms 1.1.1 2021-09-26 [1] CRAN (R 4.1.2)
htmlTable 2.4.0 2022-01-04 [1] CRAN (R 4.1.0)
htmltools 0.5.2 2021-08-25 [1] CRAN (R 4.1.2)
htmlwidgets 1.5.4 2021-09-08 [1] CRAN (R 4.1.0)
httpuv 1.6.5 2022-01-05 [1] CRAN (R 4.1.2)
httr 1.4.2 2020-07-20 [1] CRAN (R 4.1.2)
IRanges * 2.28.0 2021-10-26 [1] Bioconductor
iterators * 1.0.14 2022-02-05 [1] CRAN (R 4.1.0)
jpeg 0.1-9 2021-07-24 [1] CRAN (R 4.1.0)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.1.2)
jsonlite 1.7.3 2022-01-17 [1] CRAN (R 4.1.2)
KEGGREST 1.34.0 2021-10-26 [1] Bioconductor
knitr 1.37 2021-12-16 [1] CRAN (R 4.1.0)
labeling 0.4.2 2020-10-20 [1] CRAN (R 4.1.2)
later 1.3.0 2021-08-18 [1] CRAN (R 4.1.0)
lattice 0.20-45 2021-09-22 [2] CRAN (R 4.1.2)
latticeExtra 0.6-29 2019-12-19 [1] CRAN (R 4.1.0)
lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.1.0)
lifecycle 1.0.1 2021-09-24 [1] CRAN (R 4.1.2)
magrittr 2.0.2 2022-01-26 [1] CRAN (R 4.1.2)
Matrix 1.4-0 2021-12-08 [1] CRAN (R 4.1.2)
MatrixGenerics * 1.6.0 2021-10-26 [1] Bioconductor
matrixStats * 0.61.0 2021-09-17 [1] CRAN (R 4.1.2)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.1.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.1.2)
nnet 7.3-16 2021-05-03 [2] CRAN (R 4.1.2)
pillar 1.6.5 2022-01-25 [1] CRAN (R 4.1.2)
pkgbuild 1.3.1 2021-12-20 [1] CRAN (R 4.1.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.1.2)
pkgload 1.2.4 2021-11-30 [1] CRAN (R 4.1.0)
plotly 4.10.0 2021-10-09 [1] CRAN (R 4.1.0)
plyr 1.8.6 2020-03-03 [1] CRAN (R 4.1.0)
png 0.1-7 2013-12-03 [1] CRAN (R 4.1.0)
prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.1.2)
processx 3.5.2 2021-04-30 [1] CRAN (R 4.1.2)
progress 1.2.2 2019-05-16 [1] CRAN (R 4.1.2)
promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.1.0)
ProtGenerics 1.26.0 2021-10-26 [1] Bioconductor
ps 1.6.0 2021-02-28 [1] CRAN (R 4.1.2)
purrr 0.3.4 2020-04-17 [1] CRAN (R 4.1.2)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.1.2)
rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.1.2)
RColorBrewer 1.1-2 2014-12-07 [1] CRAN (R 4.1.2)
Rcpp 1.0.8 2022-01-13 [1] CRAN (R 4.1.2)
RCurl 1.98-1.5 2021-09-17 [1] CRAN (R 4.1.0)
readxl * 1.3.1 2019-03-13 [1] CRAN (R 4.1.2)
remotes 2.4.2 2021-11-30 [1] CRAN (R 4.1.0)
reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.1.0)
restfulr 0.0.13 2017-08-06 [1] CRAN (R 4.1.0)
rjson 0.2.21 2022-01-09 [1] CRAN (R 4.1.0)
rlang 1.0.0 2022-01-26 [1] CRAN (R 4.1.2)
rmarkdown 2.11 2021-09-14 [1] CRAN (R 4.1.2)
rpart 4.1-15 2019-04-12 [2] CRAN (R 4.1.2)
rprojroot 2.0.2 2020-11-15 [1] CRAN (R 4.1.0)
Rsamtools 2.10.0 2021-10-26 [1] Bioconductor
RSQLite 2.2.9 2021-12-06 [1] CRAN (R 4.1.0)
rstudioapi 0.13 2020-11-12 [1] CRAN (R 4.1.2)
rtracklayer 1.54.0 2021-10-26 [1] Bioconductor
S4Vectors * 0.32.3 2021-11-21 [1] Bioconductor
scales 1.1.1 2020-05-11 [1] CRAN (R 4.1.2)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.1.0)
shape 1.4.6 2021-05-19 [1] CRAN (R 4.1.0)
stringi 1.7.6 2021-11-29 [1] CRAN (R 4.1.0)
stringr 1.4.0 2019-02-10 [1] CRAN (R 4.1.0)
SummarizedExperiment * 1.24.0 2021-10-26 [1] Bioconductor
survival 3.2-13 2021-08-24 [2] CRAN (R 4.1.2)
testthat 3.1.2 2022-01-20 [1] CRAN (R 4.1.0)
tibble 3.1.6 2021-11-07 [1] CRAN (R 4.1.2)
tidyr 1.2.0 2022-02-01 [1] CRAN (R 4.1.0)
tidyselect 1.1.1 2021-04-30 [1] CRAN (R 4.1.2)
usethis 2.1.5 2021-12-09 [1] CRAN (R 4.1.0)
utf8 1.2.2 2021-07-24 [1] CRAN (R 4.1.2)
VariantAnnotation 1.40.0 2021-10-26 [1] Bioconductor
vctrs 0.3.8 2021-04-29 [1] CRAN (R 4.1.2)
viridisLite 0.4.0 2021-04-13 [1] CRAN (R 4.1.2)
withr 2.4.3 2021-11-30 [1] CRAN (R 4.1.2)
workflowr 1.7.0 2021-12-21 [1] CRAN (R 4.1.2)
xfun 0.29 2021-12-14 [1] CRAN (R 4.1.2)
XML 3.99-0.8 2021-09-17 [1] CRAN (R 4.1.0)
xml2 1.3.3 2021-11-30 [1] CRAN (R 4.1.0)
XVector 0.34.0 2021-10-26 [1] Bioconductor
yaml 2.2.2 2022-01-25 [1] CRAN (R 4.1.2)
zlibbioc 1.40.0 2021-10-26 [1] Bioconductor
[1] /mnt/beegfs/mccarthy/backed_up/general/rlyu/Software/Rlibs/4.1
[2] /opt/R/4.1.2/lib/R/library
──────────────────────────────────────────────────────────────────────────────