2022-Mar-14_permutation-based-tests-for-gendist-differences-in-groups-Fancm-ms-pvalues
Ruqian Lyu
3/14/2022
Last updated: 2022-03-14
Checks: 5 1
Knit directory: yeln_2019_spermtyping/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Tracking code development and connecting the code version to the results is critical for reproducibility. To start using Git, open the Terminal and type git init in your project directory.
This project is not being versioned with Git. To obtain the full reproducibility benefits of using workflowr, please see ?wflow_start.
Bulk BC1F1 HET versus WT
bc1f1_samples <- readRDS(file = "output/outputR/analysisRDS/all_rse_count_07-20.rds")
BiocParallel::register(MulticoreParam(workers = 12))bc1f1_samples_dist <- calGeneticDist(bc1f1_samples,group_by = "sampleGroup" )
bc1f1_samples_dist_male <- calGeneticDist(bc1f1_samples[,bc1f1_samples$sampleGroup %in%
c("Male_HET","Male_WT","Male_KO")],
group_by = "sampleGroup" )
bc1f1_samples_dist_female <- calGeneticDist(bc1f1_samples[,bc1f1_samples$sampleGroup %in%
c("Female_HET","Female_WT","Female_KO")],group_by = "sampleGroup")
(observed_male_wt_het_diff <- colSums(rowData(bc1f1_samples_dist_male)[,2][,c("Male_WT","Male_HET")])) Male_WT Male_HET
1255.786 1242.336 (observed_male_ko_wt_diff <- colSums(rowData(bc1f1_samples_dist_male)[,2][,c("Male_KO","Male_WT")])) Male_KO Male_WT
1339.082 1255.786 (colSums(rowData(bc1f1_samples_dist_female)[,2])) Female_KO Female_WT Female_HET
1438.272 1406.645 1364.624 (colSums(rowData(bc1f1_samples_dist_male)[,2])) Male_KO Male_WT Male_HET
1339.082 1255.786 1242.336 permResult_male_wt_het <- permuteDist(bc1f1_samples_dist_male[,bc1f1_samples_dist_male$sampleGroup %in% c("Male_WT","Male_HET")],
group_by = "sampleGroup",B = 1000)permResult_male_wt_het$observed_diff[1] 13.45047permute_pvals_male_wt_het <- permp(sum(permResult_male_wt_het$permutes >= permResult_male_wt_het$observed_diff),
nperm = 1000,n1 = permResult_male_wt_het$nSample[1],
permResult_male_wt_het$nSample[2],
twosided = FALSE)
permute_pvals_male_wt_het [1] 0.4385614ggplot()+geom_histogram(mapping = aes(x = permResult_male_wt_het$permutes))+
theme_bw(base_size = 18)+
geom_vline(xintercept = permResult_male_wt_het$observed_diff)+
ggtitle(paste0("BC1F1 male \n(Fancm -/ versus Fancm +/-), p: ",
round(permute_pvals_male_wt_het,2)))+
xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.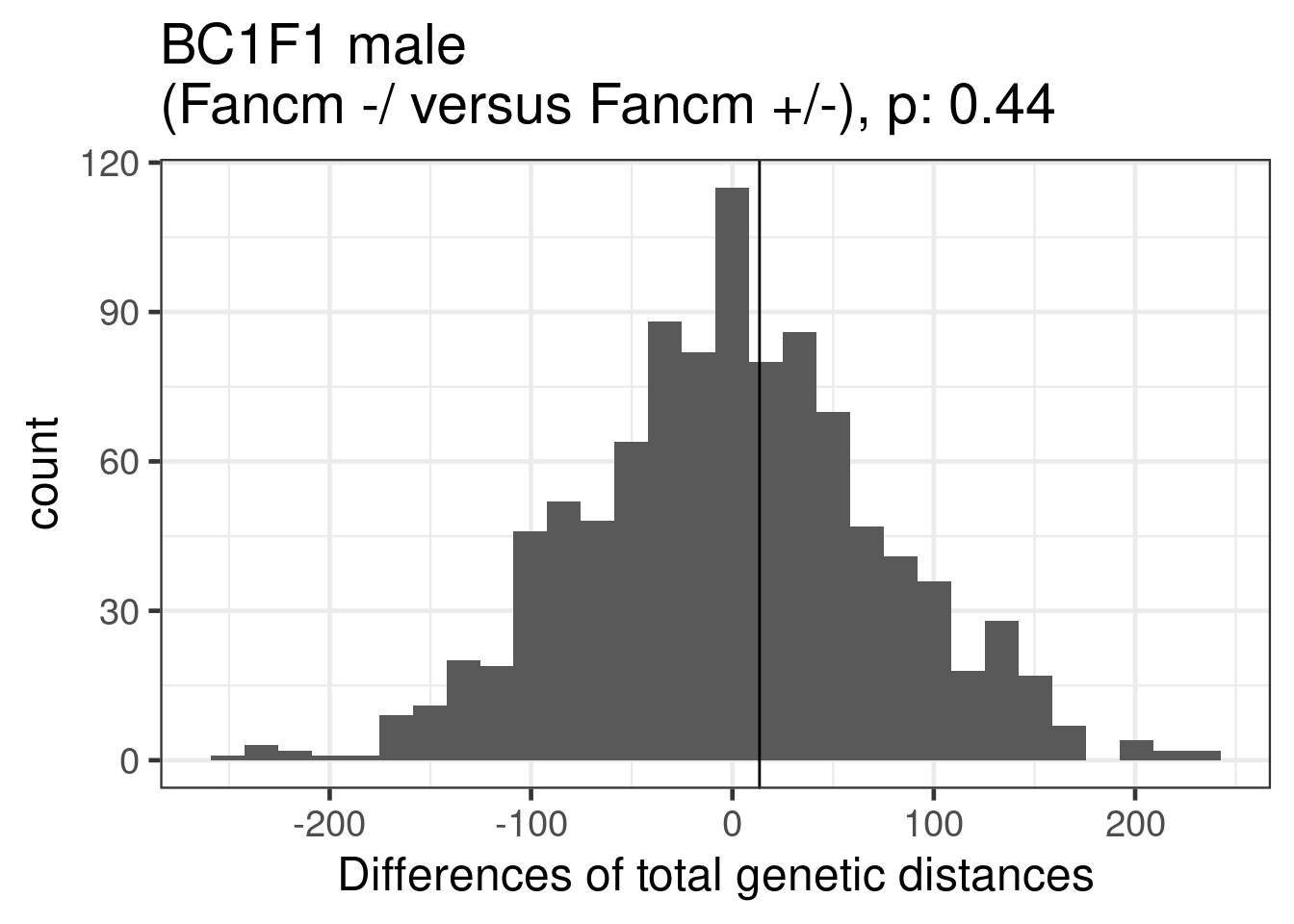
Bulk BC1F1 KO versus WT
permResult_male_ko_wt <- permuteDist(bc1f1_samples_dist_male[,bc1f1_samples_dist_male$sampleGroup %in% c("Male_KO","Male_WT")],
group_by = "sampleGroup",B = 1000)permute_pvals_male_ko_wt <- permp(sum(permResult_male_ko_wt$permutes >= permResult_male_ko_wt$observed_diff),
nperm = 1000,n1 = permResult_male_ko_wt$nSample[1],
permResult_male_ko_wt$nSample[2],twosided = FALSE)
permute_pvals_male_ko_wt [1] 0.08191808ggplot()+geom_histogram(mapping = aes(x = permResult_male_ko_wt$permutes))+theme_bw(base_size = 18)+geom_vline(xintercept = permResult_male_ko_wt$observed_diff)+ggtitle(paste0("BC1F1 male \n(Fancm -/- versus Fancm +/+), p: ", round(permute_pvals_male_ko_wt,2)))+xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.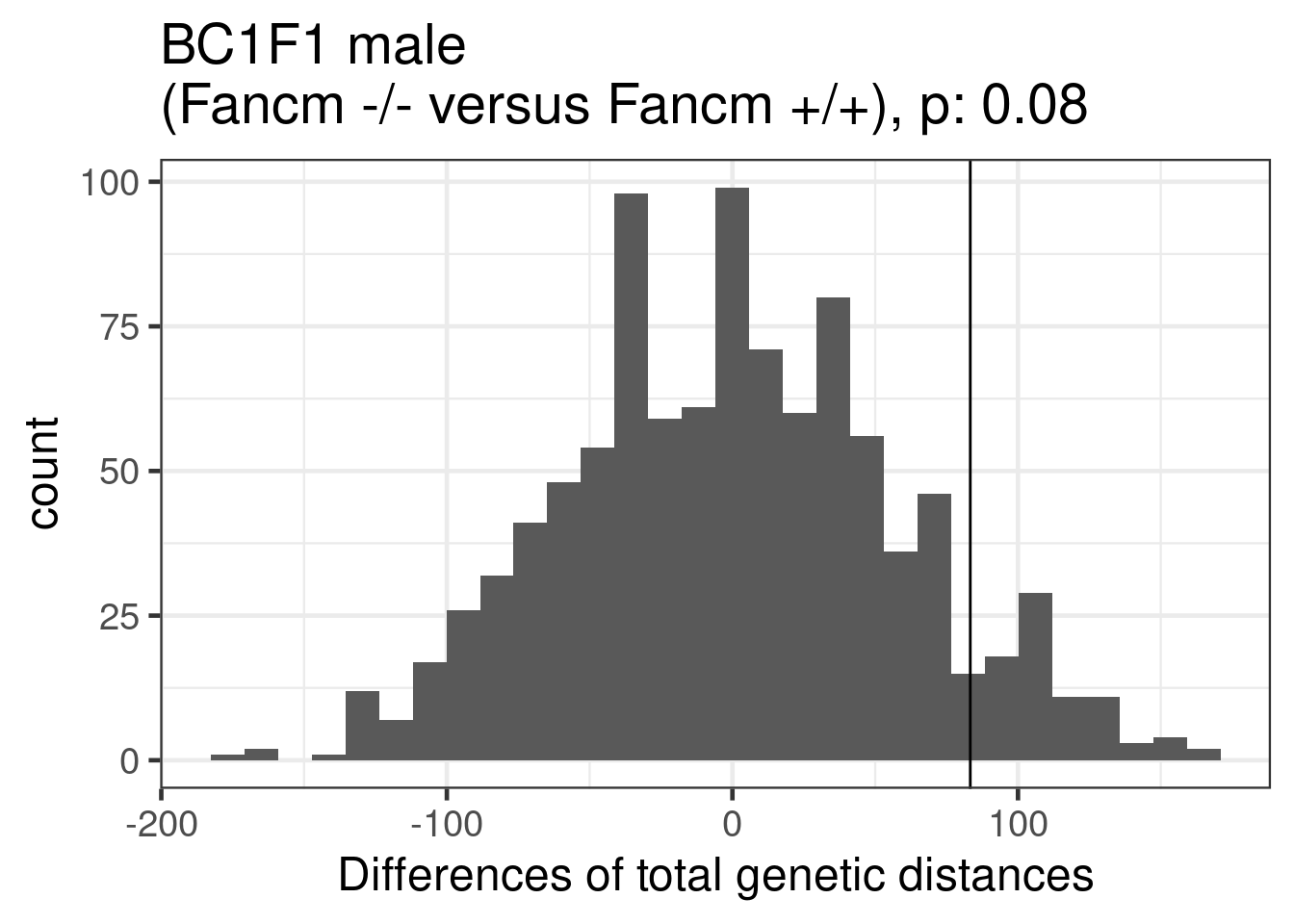
Bulk BC1F1 KO versus HET
permResult_male_ko_het <- permuteDist(bc1f1_samples_dist_male[,bc1f1_samples_dist_male$sampleGroup %in% c("Male_KO","Male_HET")],
group_by = "sampleGroup",B = 1000)permute_pvals_male_ko_het <- permp(sum(permResult_male_ko_het$permutes >= permResult_male_ko_het$observed_diff),
nperm = 1000,n1 = permResult_male_ko_het$nSample[1],
permResult_male_ko_het$nSample[2],twosided = FALSE)
permute_pvals_male_ko_het [1] 0.04095904ggplot()+geom_histogram(mapping = aes(x = permResult_male_ko_het$permutes))+theme_bw(base_size = 18)+geom_vline(xintercept = permResult_male_ko_het$observed_diff)+ggtitle(paste0("BC1F1 male \n(Fancm -/- versus Fancm +/-), p: ", round(permute_pvals_male_ko_het,2)))+xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.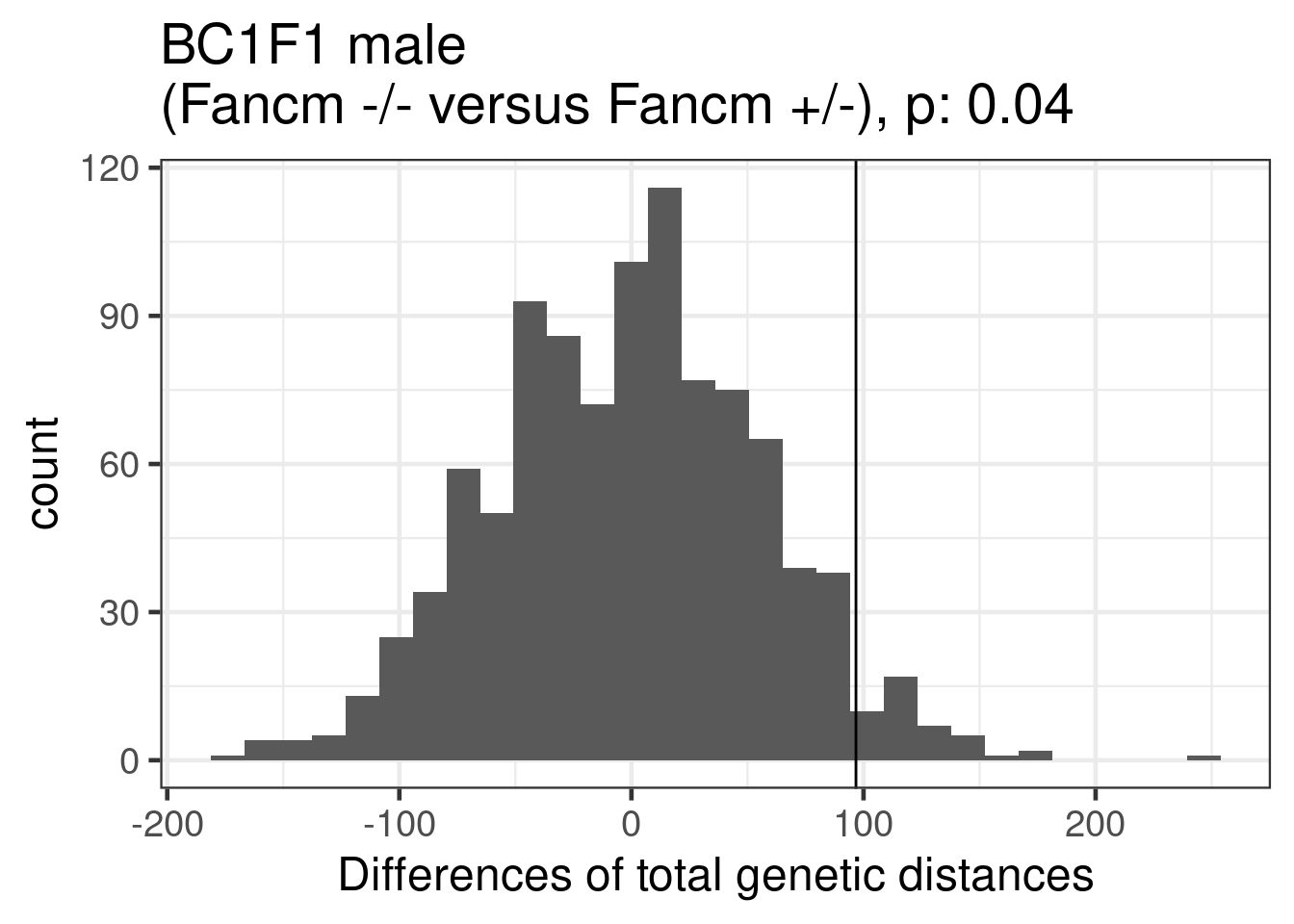
Bulk BC1F1 KO versus nonKO
bc1f1_samples_dist_male$sampleType <- plyr::mapvalues(bc1f1_samples_dist_male$sampleGroup,
from = c("Male_KO","Male_WT","Male_HET"),
to = c("Male_KO","Male_nKO","Male_nKO"))
permResult_male_ko_nko <- permuteDist(bc1f1_samples_dist_male,
group_by = "sampleType",B = 1000)permute_pvals_male_ko_nko <- permp(sum(permResult_male_ko_nko$permutes >= permResult_male_ko_nko$observed_diff),
nperm = 1000,n1 = permResult_male_ko_nko$nSample[1],
permResult_male_ko_nko$nSample[2],twosided = FALSE)
permute_pvals_male_ko_nko [1] 0.03496503ggplot()+geom_histogram(mapping = aes(x = permResult_male_ko_nko$permutes))+
theme_bw(base_size = 18)+
geom_vline(xintercept = permResult_male_ko_nko$observed_diff)+
ggtitle(paste0("BC1F1 male \n(Fancm -/- versus Fancm +/*), p: ", round(permute_pvals_male_ko_nko,2)))+
xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.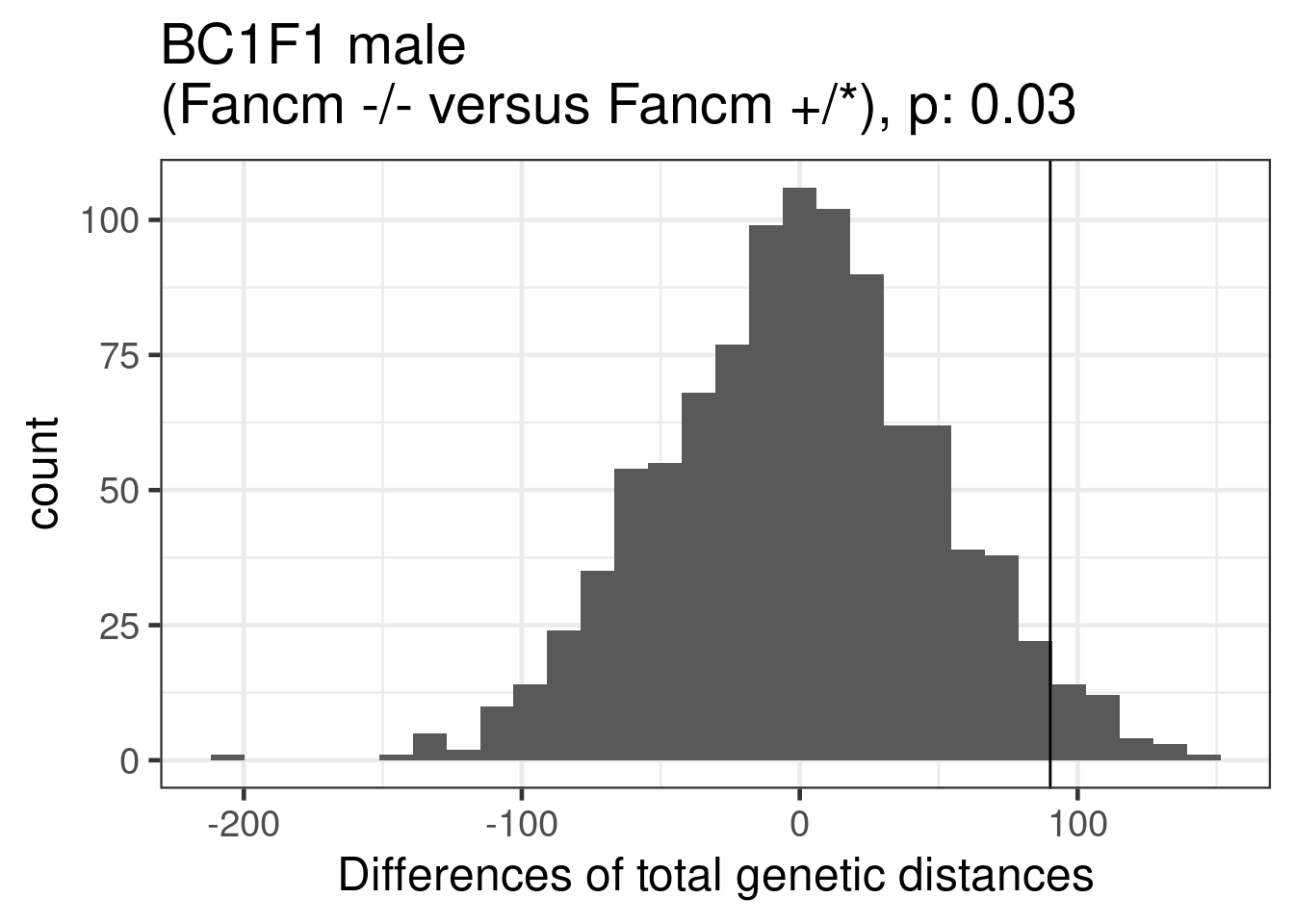 ## PCR method
## PCR method
all_rse_pcr_map <- readRDS(file ="output/outputR/analysisRDS/all_rse_pcr_map.rds")
all_rse_pcr_map$sampleGroup <- plyr::mapvalues(all_rse_pcr_map$sampleType,
from = c("Fancm-/-", "Fancm+/+"),
to = c("Mutant","Wildtype"))suppressWarnings(permResult_pcr_ko_wt <- permuteDist(all_rse_pcr_map,group_by = "sampleGroup",B=3000))permute_pvals_pcr_ko_wt <- permp(sum(permResult_pcr_ko_wt$permutes >= permResult_pcr_ko_wt$observed_diff, na.rm = T),
nperm = sum(!is.na(permResult_pcr_ko_wt$permutes)),n1 = permResult_pcr_ko_wt$nSample[1],
permResult_pcr_ko_wt$nSample[2],twosided = FALSE)ggplot()+geom_histogram(mapping = aes(x = permResult_pcr_ko_wt$permutes))+theme_bw(base_size = 18)+geom_vline(xintercept = permResult_pcr_ko_wt$observed_diff)+ggtitle(paste0("BC1F1 PCR puos male \n(Fancm -/- versus Fancm +/+), p: ", round(permute_pvals_pcr_ko_wt,3)))+xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.Warning: Removed 38 rows containing non-finite values (stat_bin).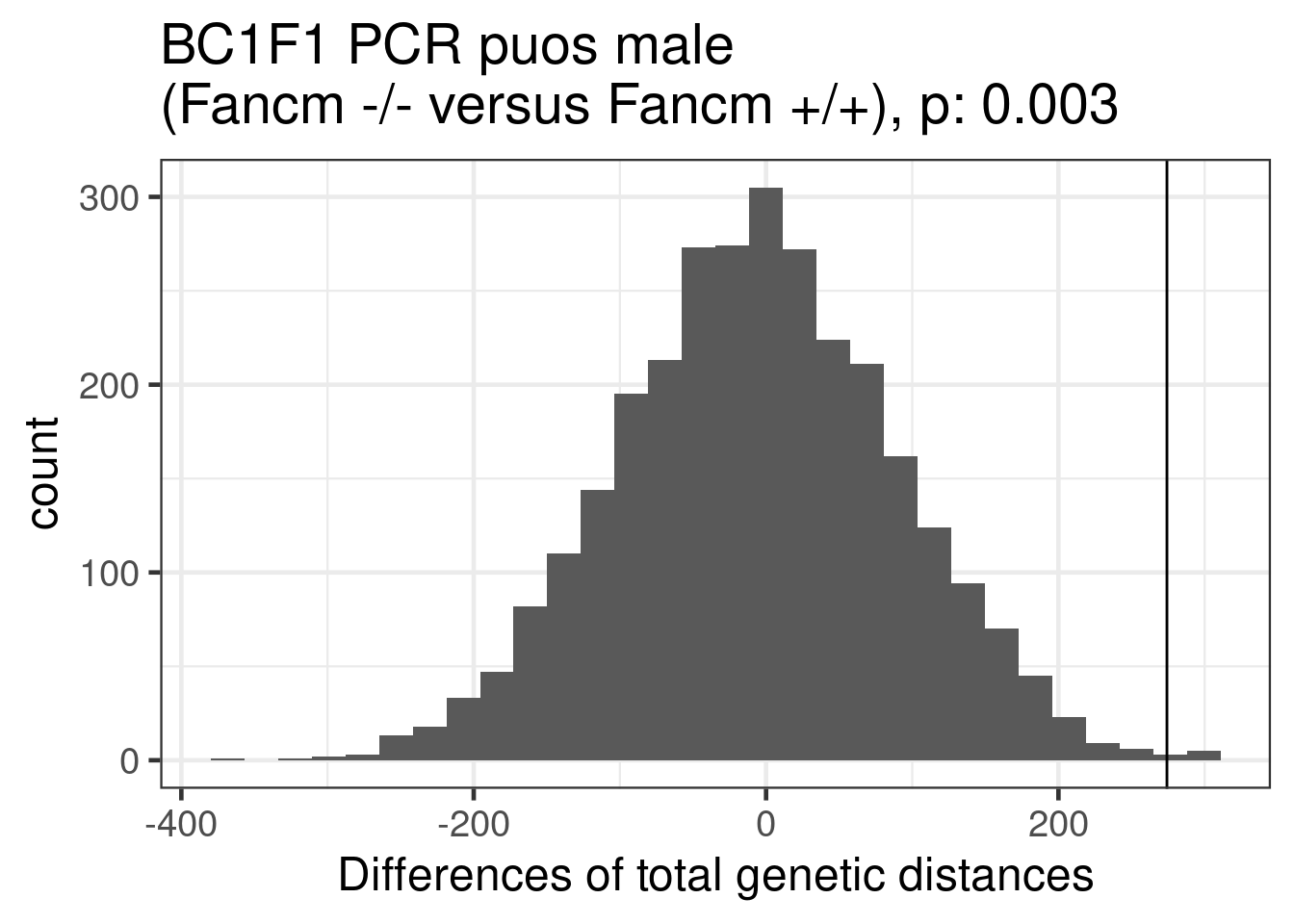
Bulk BC1F1 Female KO versus non-KO
bc1f1_samples_dist_female$sampleType <- plyr::mapvalues(bc1f1_samples_dist_female$sampleGroup,
from = c("Female_KO","Female_WT","Female_HET"),
to = c("Female_KO","Female_nKO","Female_nKO"))
permResult_female_ko_nko <- permuteDist(bc1f1_samples_dist_female,
group_by = "sampleType",B = 1000)bulk_bc1f1_female_ko_nko <- calGeneticDist(bc1f1_samples_dist_female,group_by = "sampleType")
colSums(rowData(bulk_bc1f1_female_ko_nko)[,2]) Female_KO Female_nKO
1438.272 1385.232 permute_pvals_female_ko_nko <- permp(sum(permResult_female_ko_nko$permutes >= permResult_female_ko_nko$observed_diff),
nperm = 1000,n1 = permResult_female_ko_nko$nSample[1],
permResult_female_ko_nko$nSample[2],twosided = FALSE)
permute_pvals_female_ko_nko [1] 0.1468531ggplot()+geom_histogram(mapping = aes(x = permResult_female_ko_nko$permutes))+theme_bw(base_size = 18)+geom_vline(xintercept = permResult_female_ko_nko$observed_diff)+ggtitle(paste0("BC1F1 female \n(Fancm +/+ versus Fancm +/*), p: ", round(permute_pvals_female_ko_nko,2)))+xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.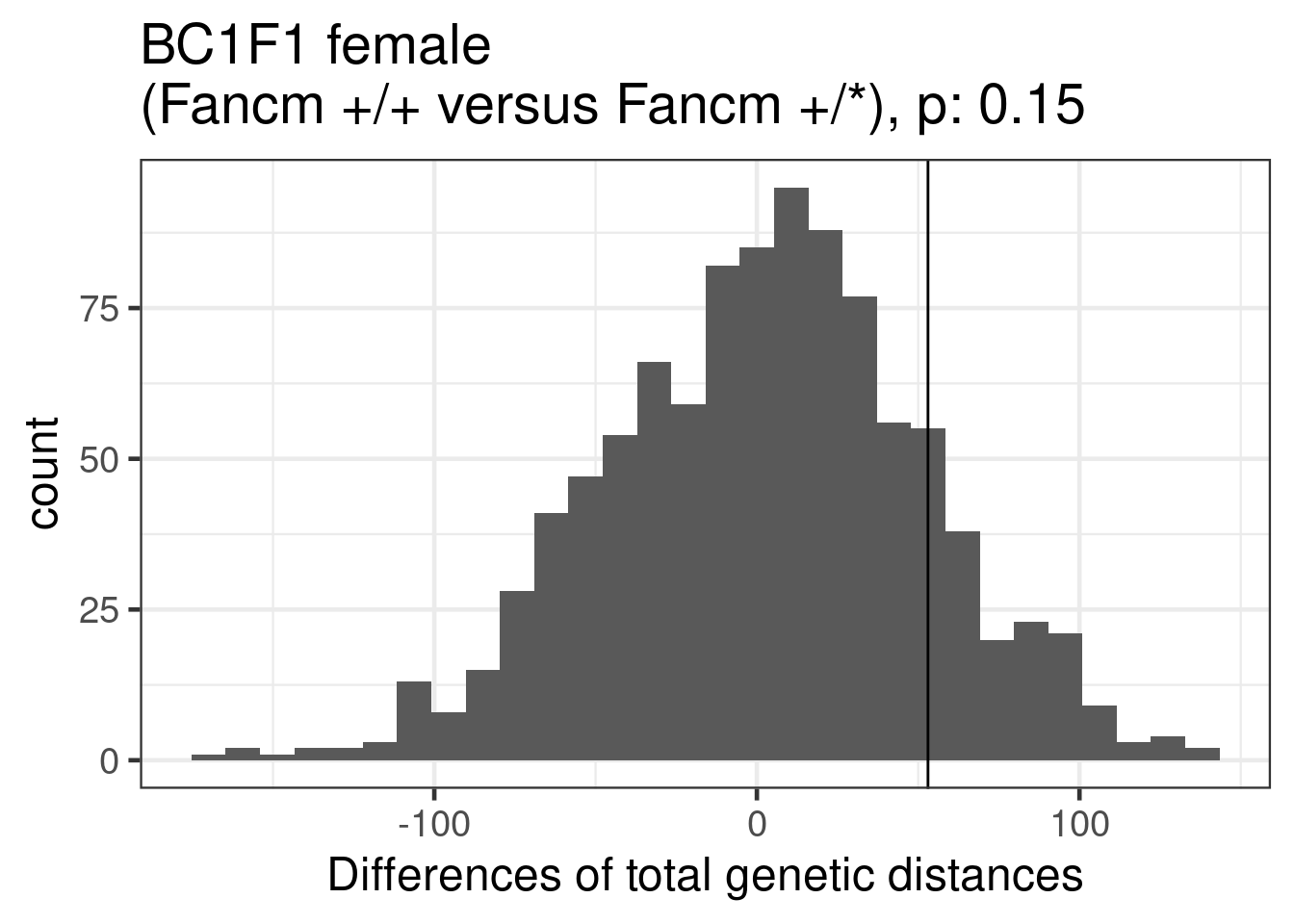
scCNV
scCNV <- readRDS(file = "~/Projects/rejy_2020_single-sperm-co-calling/output/outputR/analysisRDS/countsAll-settings4.3-scCNV-CO-counts_07-mar-2022.rds")scCNV by Fancm genotype
x <- c("mutant","mutant","wildtype","mutant",
"wildtype","wildtype")
xx <- c("Fancm-/-","Fancm-/-","Fancm+/+","Fancm-/-",
"Fancm+/+","Fancm+/+")
scCNV$sampleType <- plyr::mapvalues(scCNV$sampleGroup,from = c("WC_522",
"WC_526",
"WC_CNV_42",
"WC_CNV_43",
"WC_CNV_44",
"WC_CNV_53"),
to =x)
scCNV_dist_type <- calGeneticDist(scCNV,group_by = "sampleType")
colSums(as.matrix(rowData(scCNV_dist_type)$kosambi)) mutant wildtype
1387.336 1227.412 permResult_sccnv_ko_wt <- permuteDist(scCNV,
group_by = "sampleType",B = 1000)permute_pvals_scCNV_ko_wt <- permp(sum(permResult_sccnv_ko_wt$permutes >= permResult_sccnv_ko_wt$observed_diff),
nperm = 1000,n1 = permResult_sccnv_ko_wt$nSample[1],
permResult_sccnv_ko_wt$nSample[2],twosided = FALSE)
permute_pvals_scCNV_ko_wt [1] 0.000999001ggplot()+geom_histogram(mapping = aes(x = permResult_sccnv_ko_wt$permutes))+theme_bw(base_size = 18)+geom_vline(xintercept = permResult_sccnv_ko_wt$observed_diff)+ggtitle(paste0("F1 single sperm sequencing \n(Fancm +/+ versus Fancm +/-), p: ", round(permute_pvals_scCNV_ko_wt,3)))+xlab("Differences of total genetic distances")`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.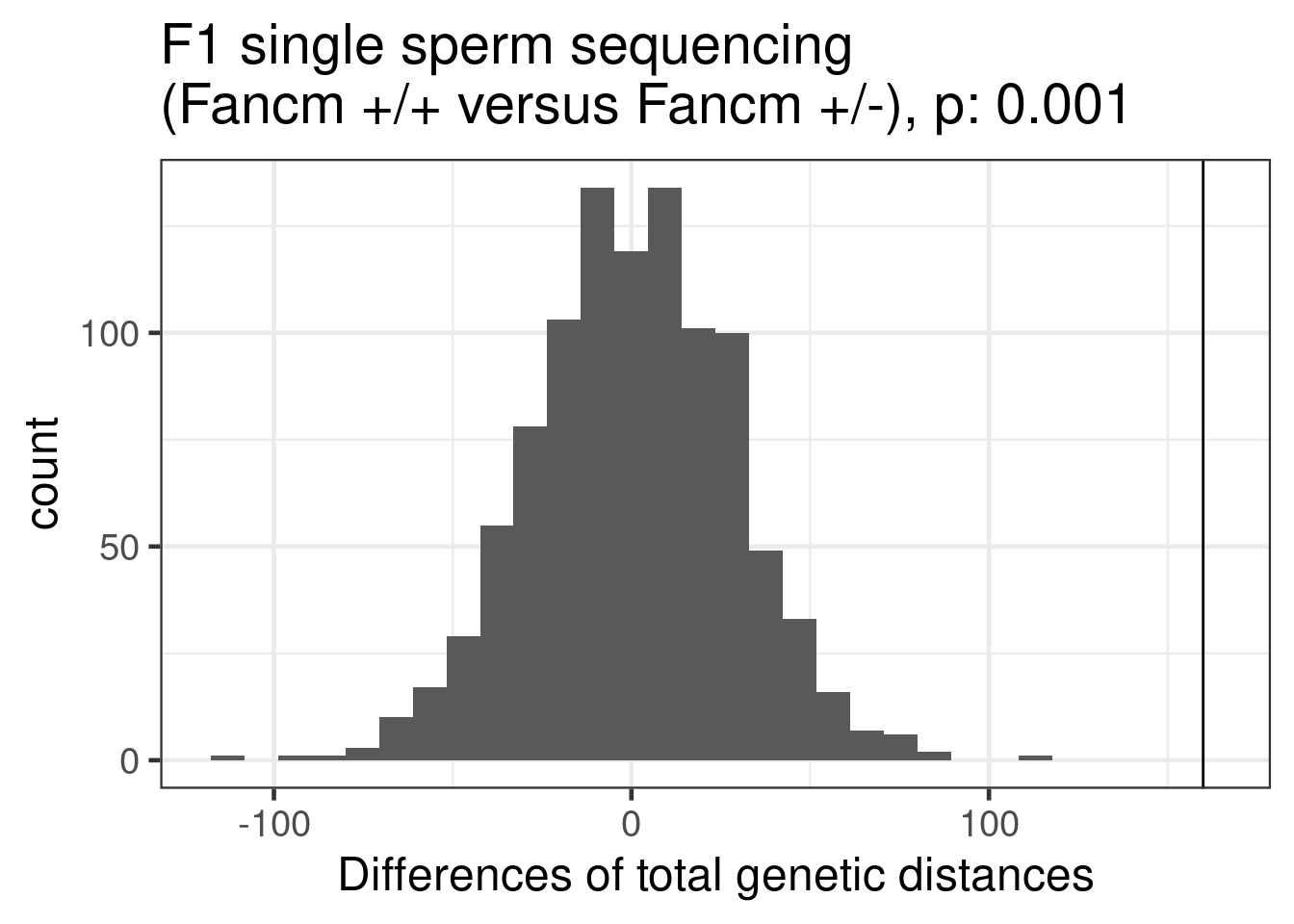
Chromosome difference
permuteSampleType <- function(co_count, B = 1000,
permuteCol = "sampleType"){
len_1 <- table(colData(co_count)[,permuteCol])[1]
permutedCoCount <- co_count
bbl <- bptry(bplapply(1:B, function(x){
type1Idx <- sample(seq(ncol(permutedCoCount)),len_1)
type2Idx <- setdiff(seq(ncol(permutedCoCount)),type1Idx )
stopifnot(length(type1Idx)>0)
stopifnot(length(type2Idx)>0)
# sink(NULL, type = "message")
# message("type1Idx",paste0(type1Idx,collapse = ","),"\n")
# message("type2Idx",paste0(type2Idx,collapse = ","),"\n")
#
#Sys.sleep(3)
permutedCoCount$sampleType[type1Idx] <- names(table(permutedCoCount$sampleType))[1]
permutedCoCount$sampleType[type2Idx] <- names(table(permutedCoCount$sampleType))[2]
permutedCoCount_dist_bin_dist <- calGeneticDist(permutedCoCount,group_by = permuteCol)
mcols(permutedCoCount_dist_bin_dist)[,2]
}),bplist_error=identity)
observed_chr_dist_diff <- calGeneticDist(co_count,group_by = permuteCol)
observed_chr_dist_diff <- bplapply(paste0("chr",1:19), function(chr){
tmp <- observed_chr_dist_diff[seqnames(observed_chr_dist_diff) ==chr,]
c("chrom"=chr,colSums(rowData(tmp)[,2]),
"diff" = (colSums(rowData(tmp)[,2])[1] - colSums(rowData(tmp)[,2])[2]) )})
observed_dist_bin_diff <- do.call(rbind,observed_chr_dist_diff)
mt_scnv_permute <- sapply(bbl,function(x){
lapply(paste0("chr",1:19), function(chr){
tmp <- x[as.character(seqnames(co_count))==chr,]
c("chrom"= chr,colSums(tmp),
"diff" = (colSums(tmp)["mutant"] -colSums(tmp)["wildtype"]))})
})
permute_statistic <- data.frame(do.call(rbind,mt_scnv_permute))
colnames(permute_statistic) <- c("chrom","wildtype","mutant","diff")
permute_statistic$diff <- as.numeric(permute_statistic$diff)
observed_dist_bin_diff <- data.frame(observed_dist_bin_diff)
observed_dist_bin_diff$diff.mutant <- as.numeric(observed_dist_bin_diff$diff.mutant)
p <- permute_statistic %>% dplyr::left_join(observed_dist_bin_diff,by ="chrom") %>%
mutate(diff.mutant = as.numeric(diff.mutant)) %>% ggplot()+ geom_histogram(mapping = aes(x = diff)) +
geom_vline(mapping = aes(xintercept=diff.mutant)) +facet_wrap(.~chrom)
permute_statistic_agg <- lapply(paste0("chr",1:19), function(chr){
permute_statistic %>% filter(chrom ==chr) %>%
summarise(extrtimes = sum(diff >= (observed_dist_bin_diff$diff.mutant[observed_dist_bin_diff$chrom==chr])),
chrom = chr)
})
permute_statistic_agg <- do.call(rbind, permute_statistic_agg)
permute_pvals <- permp(permute_statistic_agg[,1],nperm = B,n1 =len_1,
n2 = (ncol(co_count)-len_1),twosided = FALSE)
temp_gr <- cbind(permute_statistic_agg,pval = permute_pvals)
list(p_val = temp_gr,
plot = p)
}permuteCol <- "sampleType"
permute_pvals <- permuteSampleType(scCNV, B=1000)
permute_pvals$p_val extrtimes chrom pval
1 3 chr1 0.003996004
2 13 chr2 0.013986014
3 546 chr3 0.546453546
4 1 chr4 0.001998002
5 389 chr5 0.389610390
6 311 chr6 0.311688312
7 143 chr7 0.143856144
8 2 chr8 0.002997003
9 28 chr9 0.028971029
10 610 chr10 0.610389610
11 5 chr11 0.005994006
12 549 chr12 0.549450549
13 7 chr13 0.007992008
14 588 chr14 0.588411588
15 110 chr15 0.110889111
16 385 chr16 0.385614386
17 99 chr17 0.099900100
18 27 chr18 0.027972028
19 70 chr19 0.070929071#permute_pvalspermute_pvals$p_val[permute_pvals$p_val$pval<0.05,] extrtimes chrom pval
1 3 chr1 0.003996004
2 13 chr2 0.013986014
4 1 chr4 0.001998002
8 2 chr8 0.002997003
9 28 chr9 0.028971029
11 5 chr11 0.005994006
13 7 chr13 0.007992008
18 27 chr18 0.027972028permute_pvals$plot`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.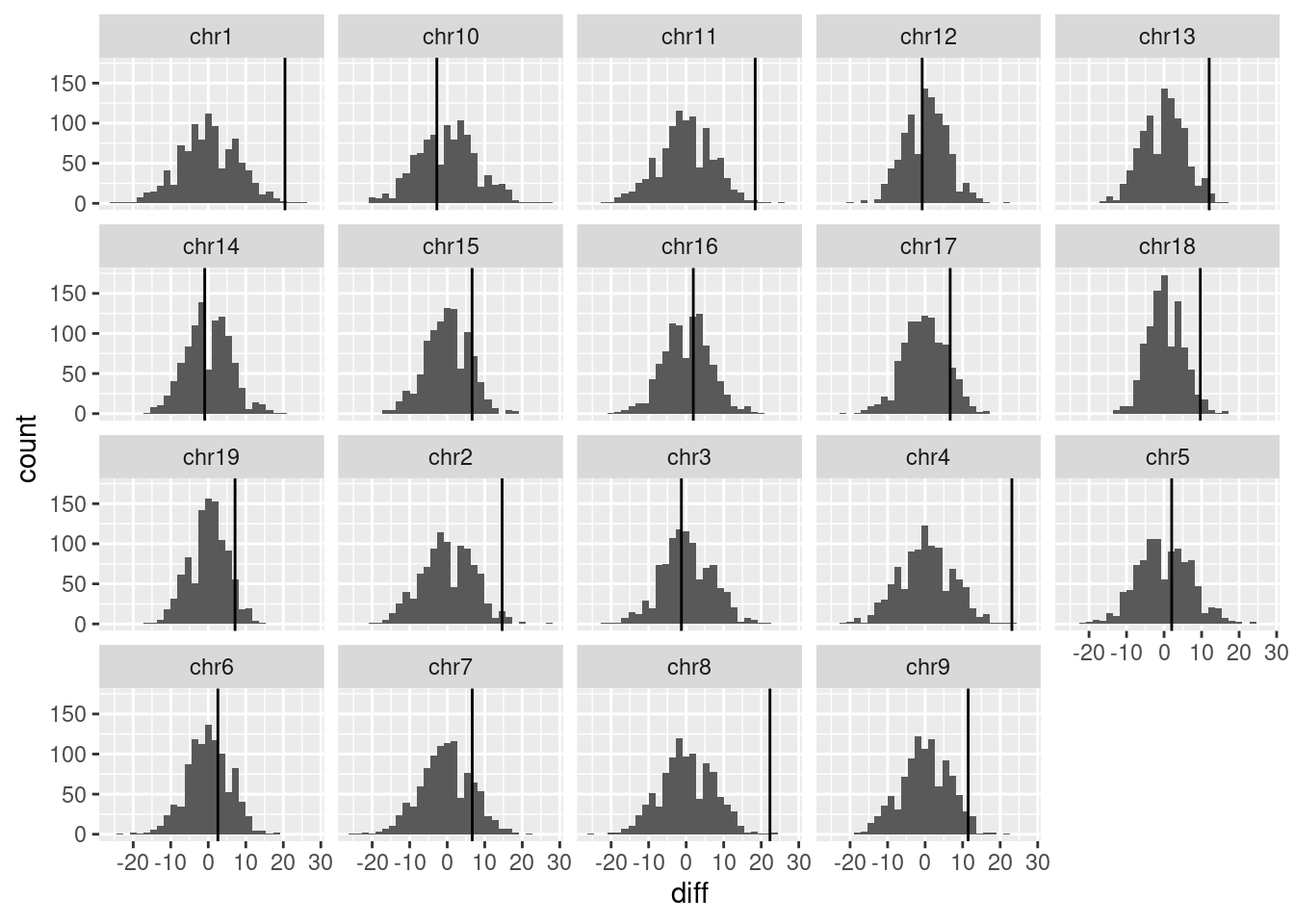
#pMultiple testing correction
padj <- cbind(permute_pvals$p_val, fdr = p.adjust(permute_pvals$p_val$pval,method = "fdr"))
padj extrtimes chrom pval fdr
1 3 chr1 0.003996004 0.02530803
2 13 chr2 0.013986014 0.04428904
3 546 chr3 0.546453546 0.61038961
4 1 chr4 0.001998002 0.02530803
5 389 chr5 0.389610390 0.49350649
6 311 chr6 0.311688312 0.45554446
7 143 chr7 0.143856144 0.22777223
8 2 chr8 0.002997003 0.02530803
9 28 chr9 0.028971029 0.06880619
10 610 chr10 0.610389610 0.61038961
11 5 chr11 0.005994006 0.02847153
12 549 chr12 0.549450549 0.61038961
13 7 chr13 0.007992008 0.03036963
14 588 chr14 0.588411588 0.61038961
15 110 chr15 0.110889111 0.19153574
16 385 chr16 0.385614386 0.49350649
17 99 chr17 0.099900100 0.18981019
18 27 chr18 0.027972028 0.06880619
19 70 chr19 0.070929071 0.14973915padj[padj$fdr<0.055,] extrtimes chrom pval fdr
1 3 chr1 0.003996004 0.02530803
2 13 chr2 0.013986014 0.04428904
4 1 chr4 0.001998002 0.02530803
8 2 chr8 0.002997003 0.02530803
11 5 chr11 0.005994006 0.02847153
13 7 chr13 0.007992008 0.03036963
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.1.2 (2021-11-01)
os Rocky Linux 8.5 (Green Obsidian)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2022-03-14
pandoc 2.11.4 @ /usr/lib/rstudio-server/bin/pandoc/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
AnnotationDbi 1.56.2 2021-11-09 [1] Bioconductor
AnnotationFilter 1.18.0 2021-10-26 [1] Bioconductor
assertthat 0.2.1 2019-03-21 [1] CRAN (R 4.1.2)
backports 1.4.1 2021-12-13 [1] CRAN (R 4.1.2)
base64enc 0.1-3 2015-07-28 [1] CRAN (R 4.1.2)
Biobase * 2.54.0 2021-10-26 [1] Bioconductor
BiocFileCache 2.2.1 2022-01-23 [1] Bioconductor
BiocGenerics * 0.40.0 2021-10-26 [1] Bioconductor
BiocIO 1.4.0 2021-10-26 [1] Bioconductor
BiocParallel * 1.28.3 2021-12-09 [1] Bioconductor
biomaRt 2.50.3 2022-02-03 [1] Bioconductor
Biostrings 2.62.0 2021-10-26 [1] Bioconductor
biovizBase 1.42.0 2021-10-26 [1] Bioconductor
bit 4.0.4 2020-08-04 [1] CRAN (R 4.1.2)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.1.2)
bitops 1.0-7 2021-04-24 [1] CRAN (R 4.1.2)
blob 1.2.2 2021-07-23 [1] CRAN (R 4.1.2)
brio 1.1.3 2021-11-30 [1] CRAN (R 4.1.0)
BSgenome 1.62.0 2021-10-26 [1] Bioconductor
cachem 1.0.6 2021-08-19 [1] CRAN (R 4.1.0)
callr 3.7.0 2021-04-20 [1] CRAN (R 4.1.2)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.1.2)
checkmate 2.0.0 2020-02-06 [1] CRAN (R 4.1.0)
circlize 0.4.13 2021-06-09 [1] CRAN (R 4.1.0)
cli 3.1.1 2022-01-20 [1] CRAN (R 4.1.2)
cluster 2.1.2 2021-04-17 [2] CRAN (R 4.1.2)
codetools 0.2-18 2020-11-04 [2] CRAN (R 4.1.2)
colorspace 2.0-2 2021-06-24 [1] CRAN (R 4.1.2)
comapr * 0.99.43 2022-03-09 [1] Github (ruqianl/comapr@915d97c)
crayon 1.4.2 2021-10-29 [1] CRAN (R 4.1.2)
curl 4.3.2 2021-06-23 [1] CRAN (R 4.1.2)
data.table 1.14.2 2021-09-27 [1] CRAN (R 4.1.2)
DBI 1.1.2 2021-12-20 [1] CRAN (R 4.1.2)
dbplyr 2.1.1 2021-04-06 [1] CRAN (R 4.1.2)
DelayedArray 0.20.0 2021-10-26 [1] Bioconductor
desc 1.4.0 2021-09-28 [1] CRAN (R 4.1.0)
devtools 2.4.3 2021-11-30 [1] CRAN (R 4.1.0)
dichromat 2.0-0 2013-01-24 [1] CRAN (R 4.1.0)
digest 0.6.29 2021-12-01 [1] CRAN (R 4.1.2)
dplyr * 1.0.7 2021-06-18 [1] CRAN (R 4.1.2)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.1.2)
ensembldb 2.18.3 2022-01-13 [1] Bioconductor
evaluate 0.14 2019-05-28 [1] CRAN (R 4.1.2)
fansi 1.0.2 2022-01-14 [1] CRAN (R 4.1.2)
farver 2.1.0 2021-02-28 [1] CRAN (R 4.1.2)
fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.1.2)
filelock 1.0.2 2018-10-05 [1] CRAN (R 4.1.0)
foreach 1.5.2 2022-02-02 [1] CRAN (R 4.1.0)
foreign 0.8-81 2020-12-22 [2] CRAN (R 4.1.2)
Formula 1.2-4 2020-10-16 [1] CRAN (R 4.1.0)
fs 1.5.2 2021-12-08 [1] CRAN (R 4.1.2)
generics 0.1.1 2021-10-25 [1] CRAN (R 4.1.2)
GenomeInfoDb * 1.30.1 2022-01-30 [1] Bioconductor
GenomeInfoDbData 1.2.7 2022-01-28 [1] Bioconductor
GenomicAlignments 1.30.0 2021-10-26 [1] Bioconductor
GenomicFeatures 1.46.4 2022-01-20 [1] Bioconductor
GenomicRanges * 1.46.1 2021-11-18 [1] Bioconductor
ggplot2 * 3.3.5 2021-06-25 [1] CRAN (R 4.1.2)
git2r 0.29.0 2021-11-22 [1] CRAN (R 4.1.2)
GlobalOptions 0.1.2 2020-06-10 [1] CRAN (R 4.1.0)
glue 1.6.1 2022-01-22 [1] CRAN (R 4.1.2)
gridExtra * 2.3 2017-09-09 [1] CRAN (R 4.1.0)
gtable 0.3.0 2019-03-25 [1] CRAN (R 4.1.2)
Gviz 1.38.3 2022-01-23 [1] Bioconductor
highr 0.9 2021-04-16 [1] CRAN (R 4.1.2)
Hmisc 4.6-0 2021-10-07 [1] CRAN (R 4.1.0)
hms 1.1.1 2021-09-26 [1] CRAN (R 4.1.2)
htmlTable 2.4.0 2022-01-04 [1] CRAN (R 4.1.0)
htmltools 0.5.2 2021-08-25 [1] CRAN (R 4.1.2)
htmlwidgets 1.5.4 2021-09-08 [1] CRAN (R 4.1.0)
httpuv 1.6.5 2022-01-05 [1] CRAN (R 4.1.2)
httr 1.4.2 2020-07-20 [1] CRAN (R 4.1.2)
IRanges * 2.28.0 2021-10-26 [1] Bioconductor
iterators 1.0.14 2022-02-05 [1] CRAN (R 4.1.0)
jpeg 0.1-9 2021-07-24 [1] CRAN (R 4.1.0)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.1.2)
jsonlite 1.7.3 2022-01-17 [1] CRAN (R 4.1.2)
KEGGREST 1.34.0 2021-10-26 [1] Bioconductor
knitr 1.37 2021-12-16 [1] CRAN (R 4.1.0)
labeling 0.4.2 2020-10-20 [1] CRAN (R 4.1.2)
later 1.3.0 2021-08-18 [1] CRAN (R 4.1.0)
lattice 0.20-45 2021-09-22 [2] CRAN (R 4.1.2)
latticeExtra 0.6-29 2019-12-19 [1] CRAN (R 4.1.0)
lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.1.0)
lifecycle 1.0.1 2021-09-24 [1] CRAN (R 4.1.2)
magrittr 2.0.2 2022-01-26 [1] CRAN (R 4.1.2)
Matrix 1.4-0 2021-12-08 [1] CRAN (R 4.1.2)
MatrixGenerics * 1.6.0 2021-10-26 [1] Bioconductor
matrixStats * 0.61.0 2021-09-17 [1] CRAN (R 4.1.2)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.1.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.1.2)
nnet 7.3-16 2021-05-03 [2] CRAN (R 4.1.2)
pillar 1.6.5 2022-01-25 [1] CRAN (R 4.1.2)
pkgbuild 1.3.1 2021-12-20 [1] CRAN (R 4.1.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.1.2)
pkgload 1.2.4 2021-11-30 [1] CRAN (R 4.1.0)
plotly 4.10.0 2021-10-09 [1] CRAN (R 4.1.0)
plyr 1.8.6 2020-03-03 [1] CRAN (R 4.1.0)
png 0.1-7 2013-12-03 [1] CRAN (R 4.1.0)
prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.1.2)
processx 3.5.2 2021-04-30 [1] CRAN (R 4.1.2)
progress 1.2.2 2019-05-16 [1] CRAN (R 4.1.2)
promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.1.0)
ProtGenerics 1.26.0 2021-10-26 [1] Bioconductor
ps 1.6.0 2021-02-28 [1] CRAN (R 4.1.2)
purrr 0.3.4 2020-04-17 [1] CRAN (R 4.1.2)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.1.2)
rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.1.2)
RColorBrewer 1.1-2 2014-12-07 [1] CRAN (R 4.1.2)
Rcpp 1.0.8 2022-01-13 [1] CRAN (R 4.1.2)
RCurl 1.98-1.5 2021-09-17 [1] CRAN (R 4.1.0)
readxl * 1.3.1 2019-03-13 [1] CRAN (R 4.1.2)
remotes 2.4.2 2021-11-30 [1] CRAN (R 4.1.0)
reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.1.0)
restfulr 0.0.13 2017-08-06 [1] CRAN (R 4.1.0)
rjson 0.2.21 2022-01-09 [1] CRAN (R 4.1.0)
rlang 1.0.0 2022-01-26 [1] CRAN (R 4.1.2)
rmarkdown 2.11 2021-09-14 [1] CRAN (R 4.1.2)
rpart 4.1-15 2019-04-12 [2] CRAN (R 4.1.2)
rprojroot 2.0.2 2020-11-15 [1] CRAN (R 4.1.0)
Rsamtools 2.10.0 2021-10-26 [1] Bioconductor
RSQLite 2.2.9 2021-12-06 [1] CRAN (R 4.1.0)
rstudioapi 0.13 2020-11-12 [1] CRAN (R 4.1.2)
rtracklayer 1.54.0 2021-10-26 [1] Bioconductor
S4Vectors * 0.32.3 2021-11-21 [1] Bioconductor
scales 1.1.1 2020-05-11 [1] CRAN (R 4.1.2)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.1.0)
shape 1.4.6 2021-05-19 [1] CRAN (R 4.1.0)
statmod * 1.4.36 2021-05-10 [1] CRAN (R 4.1.2)
stringi 1.7.6 2021-11-29 [1] CRAN (R 4.1.0)
stringr 1.4.0 2019-02-10 [1] CRAN (R 4.1.0)
SummarizedExperiment * 1.24.0 2021-10-26 [1] Bioconductor
survival 3.2-13 2021-08-24 [2] CRAN (R 4.1.2)
testthat 3.1.2 2022-01-20 [1] CRAN (R 4.1.0)
tibble 3.1.6 2021-11-07 [1] CRAN (R 4.1.2)
tidyr 1.2.0 2022-02-01 [1] CRAN (R 4.1.0)
tidyselect 1.1.1 2021-04-30 [1] CRAN (R 4.1.2)
usethis 2.1.5 2021-12-09 [1] CRAN (R 4.1.0)
utf8 1.2.2 2021-07-24 [1] CRAN (R 4.1.2)
VariantAnnotation 1.40.0 2021-10-26 [1] Bioconductor
vctrs 0.3.8 2021-04-29 [1] CRAN (R 4.1.2)
viridisLite 0.4.0 2021-04-13 [1] CRAN (R 4.1.2)
withr 2.4.3 2021-11-30 [1] CRAN (R 4.1.2)
workflowr 1.7.0 2021-12-21 [1] CRAN (R 4.1.2)
xfun 0.29 2021-12-14 [1] CRAN (R 4.1.2)
XML 3.99-0.8 2021-09-17 [1] CRAN (R 4.1.0)
xml2 1.3.3 2021-11-30 [1] CRAN (R 4.1.0)
XVector 0.34.0 2021-10-26 [1] Bioconductor
yaml 2.2.2 2022-01-25 [1] CRAN (R 4.1.2)
zlibbioc 1.40.0 2021-10-26 [1] Bioconductor
[1] /mnt/beegfs/mccarthy/backed_up/general/rlyu/Software/Rlibs/4.1
[2] /opt/R/4.1.2/lib/R/library
──────────────────────────────────────────────────────────────────────────────