Lenti barcode analysis
Christina Azodi
2022-02-28
Last updated: 2022-02-28
Checks: 5 2
Knit directory: BAUH_2020_MND-single-cell/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3_Lenti/01_fasta/ | ../output/pilot3_Lenti/01_fasta |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3_Lenti/02_blast/ | ../output/pilot3_Lenti/02_blast |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3_Lenti/03_keys/ | ../output/pilot3_Lenti/03_keys |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 409be72. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .cache/
Ignored: .config/
Ignored: .snakemake/
Ignored: BAUH_2020_MND-single-cell.Rproj
Ignored: GRCh38_turboGFP-RFP_reference/
Ignored: Homo_sapiens.GRCh38.turboGFP/
Ignored: Rplots.pdf
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/pilot3_aggr-experiments.csv
Ignored: data/pilot3_donors.txt
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/2021-04-27_pilot2_nCells-per-donor.pdf
Ignored: output/2021-08-03_pilot2_nCells-per-donor.pdf
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: output/pilot2_testing/
Ignored: output/pilot3.0/
Ignored: output/pilot3.0_MN/
Ignored: output/pilot3.0_captures-separate/
Ignored: output/pilot3.0_iPSC/
Ignored: output/pilot3_Lenti/
Ignored: references/Homo_sapiens.GRCh38.turboGFP.bed
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa.fai
Ignored: references/Homo_sapiens.GRCh38.turboGFP.filtered.gtf
Ignored: references/Homo_sapiens.GRCh38.turboGFP.gtf
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Ignored: references/pilot3/
Ignored: references/test
Ignored: references/turboGFP.fa
Ignored: references/turboGFP.gtf
Ignored: references/turboRFP.fa
Ignored: workflow/rules/
Untracked files:
Untracked: cellbender.dockerfile
Untracked: hwe1e-05_maf05_vcf_stats.txt
Untracked: hwe1e-05_vcf_stats.txt
Untracked: workflow/cellbender_jobs/
Untracked: workflow/rules_cellcalling-old-for-mergedy.smk
Unstaged changes:
Modified: analysis/2022-02-24_pilot3_LentiBarcodes.Rmd
Modified: config/config_pilot3.0_MN.yml
Modified: workflow/Snakefile
Modified: workflow/rules_cellcalling.smk
Modified: workflow/rules_cellranger-merge.smk
Modified: workflow/rules_demultiplexing.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/2022-02-24_pilot3_LentiBarcodes.Rmd) and HTML (public/2022-02-24_pilot3_LentiBarcodes.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | bddd52c | cazodi | 2022-02-25 | lenti barcode analysis |
| html | bddd52c | cazodi | 2022-02-25 | lenti barcode analysis |
Goal
Generate a map between iPSC cell barcodes (Capture 5) and MN cell barcodes (Capture 4) by identifying the lenti barcode (14bp+30bp) shared by the two.
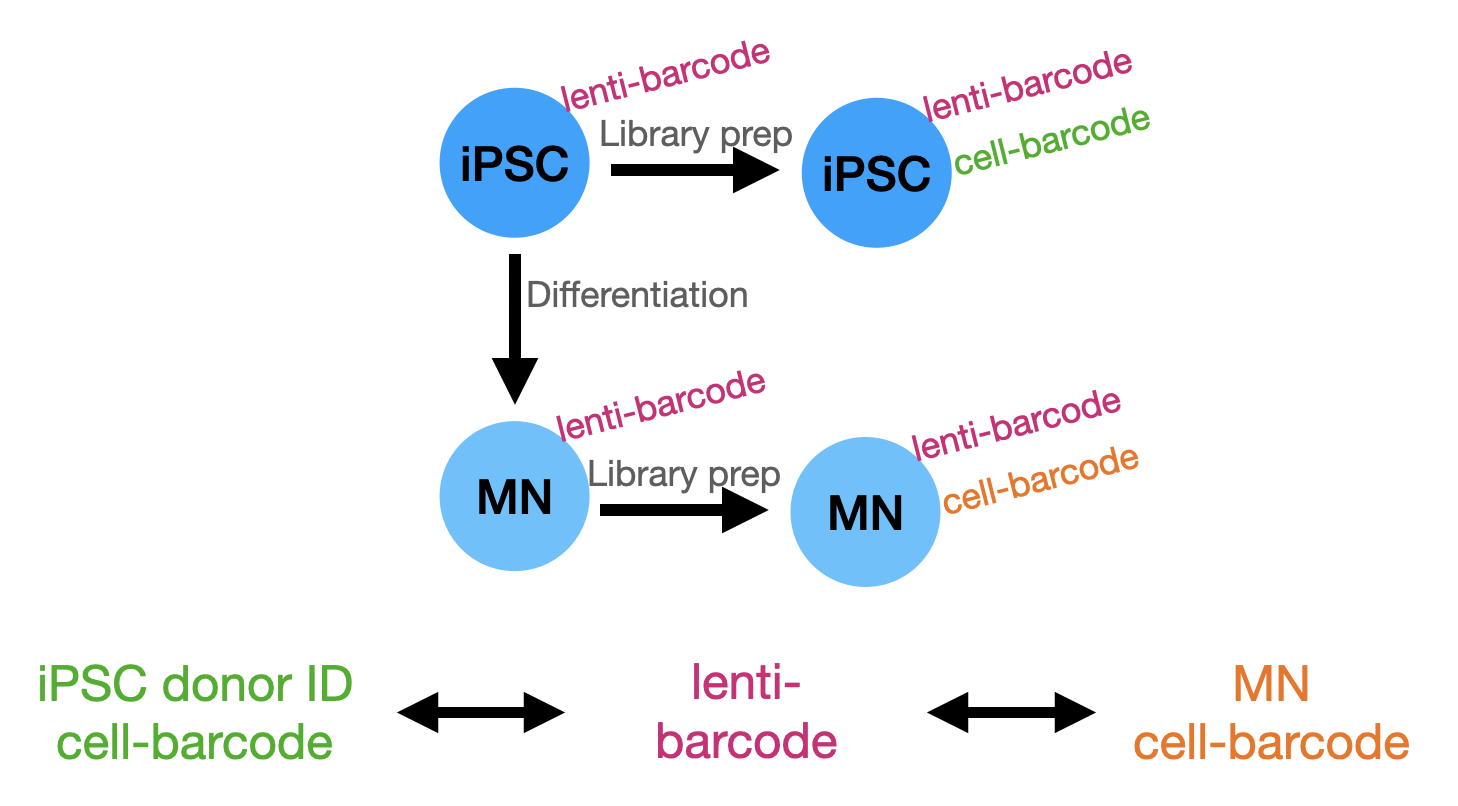
Overview of strategy
Blastn parsing

blastn strategy
Example scripts:
14 bp barcode: blastn -db output/pilot3_Lenti/00_sequences/bc14_forward.fa -query output/pilot3_Lenti/01_fasta/Capture5-Lenti.fa -query_loc 26-40 -word_size 5 -max_hsps 1 -evalue 0.1 -num_threads 4 -outfmt 6 -out output/pilot3_Lenti/02_blast/Capture5-Lenti_bc14_blast.out
30 bp barcode: blastn -db output/pilot3_Lenti/00_sequences/bc30_forward.fa -query output/pilot3_Lenti/01_fasta/Capture5-Lenti.fa -query_loc 46-76 -word_size 10 -max_hsps 1 -evalue 0.1 -max_target_seqs 1 -outfmt 6 -out output/pilot3_Lenti/02_blast/Capture5-Lenti_bc30_blast.out
Read in blastn hits and remove ambiguous matches by first keeping the longest hit for each read and if two lenti barcodes have equally long hits, removing the read entirely. Number of reads with lenti barcode matches:
load_parse_blastn <- function(path) {
df <- fread(path, header=FALSE, select = c(1:6))
names(df) <- c("qseqid", "sseqid", "pident", "length", "mismatch", "gapopen")
# If >1 hit for a lenti barcode, keep the one(s) with the longer overlap
df <- df[df[, .I[length == max(length)], by=qseqid]$V1]
# If >1 hit for a lenti barcode with equal lengths, remove them both.
df <- df[!(duplicated(df$qseqid) | duplicated(df$qseqid, fromLast = TRUE)), ]
return(df)
}
## Load and process blastn results
c4_bc14 <- load_parse_blastn(paste0(blast_out, "Capture4-Lenti_bc14_blast.out"))
c5_bc14 <- load_parse_blastn(paste0(blast_out, "Capture5-Lenti_bc14_blast.out"))
c4_bc30 <- load_parse_blastn(paste0(blast_out, "Capture4-Lenti_bc30_blast.out"))
c5_bc30 <- load_parse_blastn(paste0(blast_out, "Capture5-Lenti_bc30_blast.out"))
# Merge
c4_both <- c4_bc14 %>% inner_join(c4_bc30, by = "qseqid", suffix = c(".bc14", ".bc30"))
c5_both <- c5_bc14 %>% inner_join(c5_bc30, by = "qseqid", suffix = c(".bc14", ".bc30"))
lenti_stats <- as.data.frame(list(capture=c("Capture 4", "Capture 5"),
raw=c(31030718, 46041484),
bc14=c(nrow(c4_bc14), nrow(c5_bc14)),
bc30=c(nrow(c4_bc30), nrow(c5_bc30)),
bc14_and_30=c(nrow(c4_both), nrow(c5_both))))
rm(c4_bc14, c5_bc14, c4_bc30, c5_bc30)
lenti_stats %>% mutate_at(vars(3:5), list(percent_raw=~./raw*100)) %>%
mutate(across(where(is.numeric), round, 1)) capture raw bc14 bc30 bc14_and_30 bc14_percent_raw
1 Capture 4 31030718 5831776 2809672 2381776 18.8
2 Capture 5 46041484 17260005 12795040 12544555 37.5
bc30_percent_raw bc14_and_30_percent_raw
1 9.1 7.7
2 27.8 27.2For all the blastn hits returned (evalue <= 0.1), does the quality of the 14 bp barcode correlate to the quality of the 30 bp barcode?
pn4 <- c4_both %>% count(length.bc14) %>% mutate(length.bc14 = as.factor(length.bc14)) %>%
ggbarplot(x = "length.bc14", y = "n", label = TRUE, label.pos = "in", yscale = "log10")
pn5 <- c5_both %>% count(length.bc14) %>% mutate(length.bc14 = as.factor(length.bc14)) %>%
ggbarplot(x = "length.bc14", y = "n", label = TRUE, label.pos = "in", yscale = "log10")
p4 <- ggboxplot(c4_both, x="length.bc14", y = "length.bc30", outlier.shape = NA)
p5 <- ggboxplot(c5_both, x="length.bc14", y = "length.bc30", outlier.shape = NA)
plot_grid(pn4, pn5, p4, p5, ncol=2, rel_heights = c(1, 1.5), labels = "auto")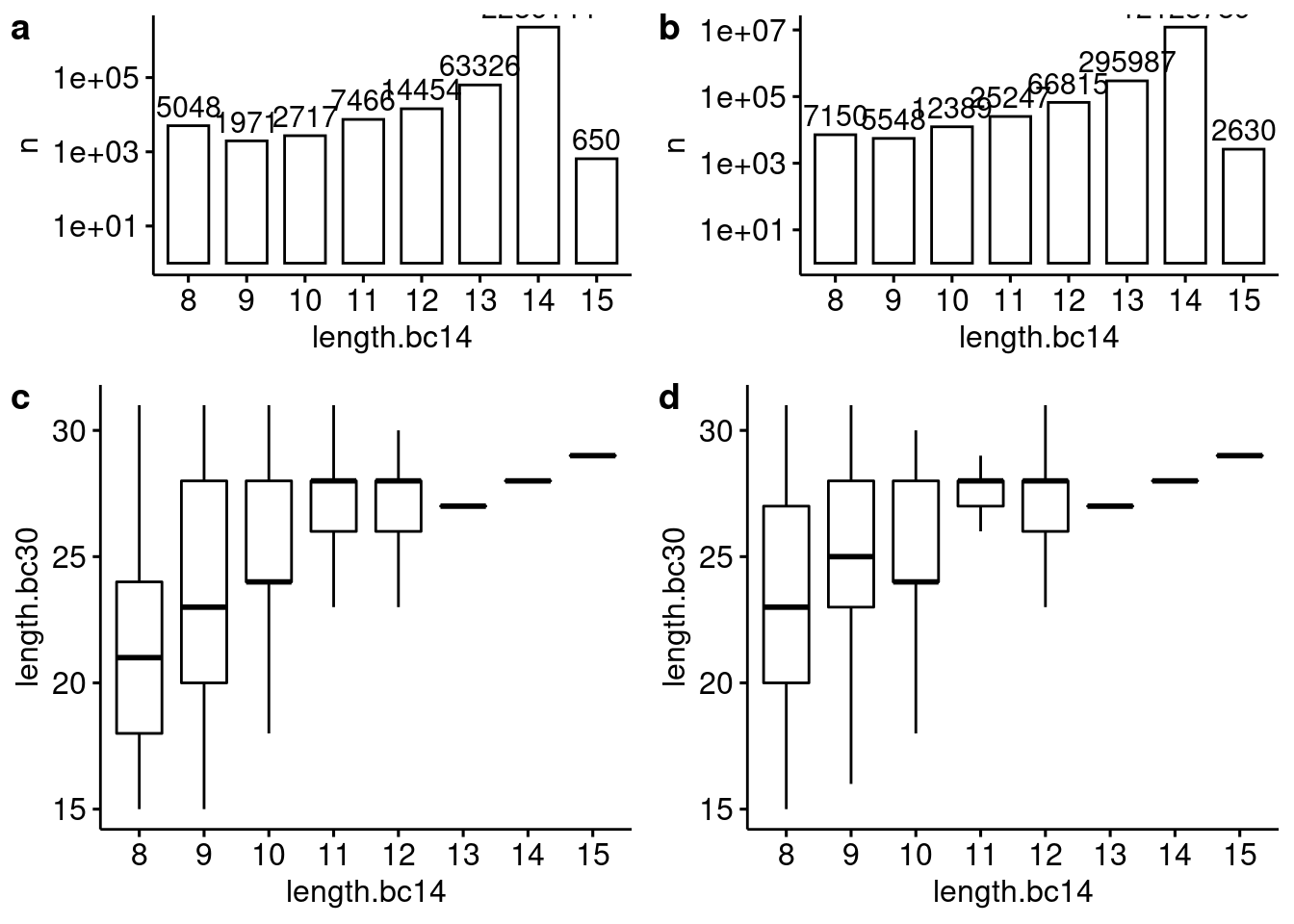
Relationship between blastn alignment length for the 14 bp and 30 bp lenti barcodes with an evalue <= 0.1 for (a) Capture4 and (b) Capture5.
Filter and merge with cell barcodes
Because we are relying primarily on the lenti barcodes for QC (as the header 25 bp and linker 4 bp regions are generally low quality), we want to set a pretty strict limit for matching. Thus we will remove any reads with lenti barcode hits <= 12 bp or 26 bp in length or with > 1 or 2 mismatches for the 14 and 30 bp barcodes, respectively.
# Add lenti barcode IDs
c4_both$lenti_bc <- paste0("lenti_", c4_both$sseqid.bc14, "-", c4_both$sseqid.bc30)
c5_both$lenti_bc <- paste0("lenti_", c5_both$sseqid.bc14, "-", c5_both$sseqid.bc30)
# Filtering by minimum length
c4_both <- subset(c4_both, c4_both$length.bc14 >= 12 & c4_both$length.bc30 >= 26)
c5_both <- subset(c5_both, c5_both$length.bc14 >= 12 & c5_both$length.bc30 >= 26)
lenti_stats$length_filter <- c(nrow(c4_both), nrow(c5_both))
# Filtering by maximum mismatches
c4_both <- subset(c4_both, c4_both$mismatch.bc14 <= 1 & c4_both$mismatch.bc30 <= 2)
c5_both <- subset(c5_both, c5_both$mismatch.bc14 <= 1 & c5_both$mismatch.bc30 <= 2)
lenti_stats$mismatch_filter <- c(nrow(c4_both), nrow(c5_both))
lenti_stats capture raw bc14 bc30 bc14_and_30 length_filter
1 Capture 4 31030718 5831776 2809672 2381776 2340970
2 Capture 5 46041484 17260005 12795040 12544555 12431016
mismatch_filter
1 2335546
2 12411301Merge with cellbarcodes from the R1 reads
The R1 reads are all exactly 28 bp and contain the 16 bp cell barcode followed by a 12 bp UMI.
parse_merge_R1_cellbarcodes <- function(path, lenti) {
cbc <- fread(path, header=FALSE, sep="\t")
summary(nchar(cbc$cell_barcode))
names(cbc) <- c("qseqid", "R1")
cbc$qseqid <- gsub(">", "", gsub(" .*", "", cbc$qseqid))
cbc <- separate(cbc, R1, into = c("cell_barcode", "umi"), sep = 16)
lenti <- lenti %>% left_join(cbc, by = "qseqid")
rm(cbc)
return(lenti)
}
c4_both <- parse_merge_R1_cellbarcodes(paste0(fasta_out, "Capture4-Lenti_R1.tsv"), c4_both)
if(save){ write.table(c4_both, paste0(out, "Capture4-Lenti_merged_barcodes.tsv"),
sep = "\t", quote=FALSE, row.names = FALSE)}
c5_both <- parse_merge_R1_cellbarcodes(paste0(fasta_out, "Capture5-Lenti_R1.tsv"), c5_both)
if(save){ write.table(c5_both, paste0(out, "Capture5-Lenti_merged_barcodes.tsv"),
sep = "\t", quote=FALSE, row.names = FALSE)}Find unambiguous cell-to-lenti barcode pairs
Ideally, each cellbarcode should be associated with a single lenti-barcode, however some cell barcodes have umis that are assigned to more than one lenti-barcode:
c4_barcode_umi_counts <- c4_both %>% distinct(cell_barcode, lenti_bc, umi) %>%
group_by(cell_barcode, lenti_bc) %>%
tally() %>% group_by(cell_barcode) %>%
mutate(percent_umis_with_lentiBC = n/sum(n),
total_umis = sum(n))
plot_pCells_c4 <- ggscatterhist(c4_barcode_umi_counts, x="n",
y="percent_umis_with_lentiBC",
color="total_umis", xscale = "log10") 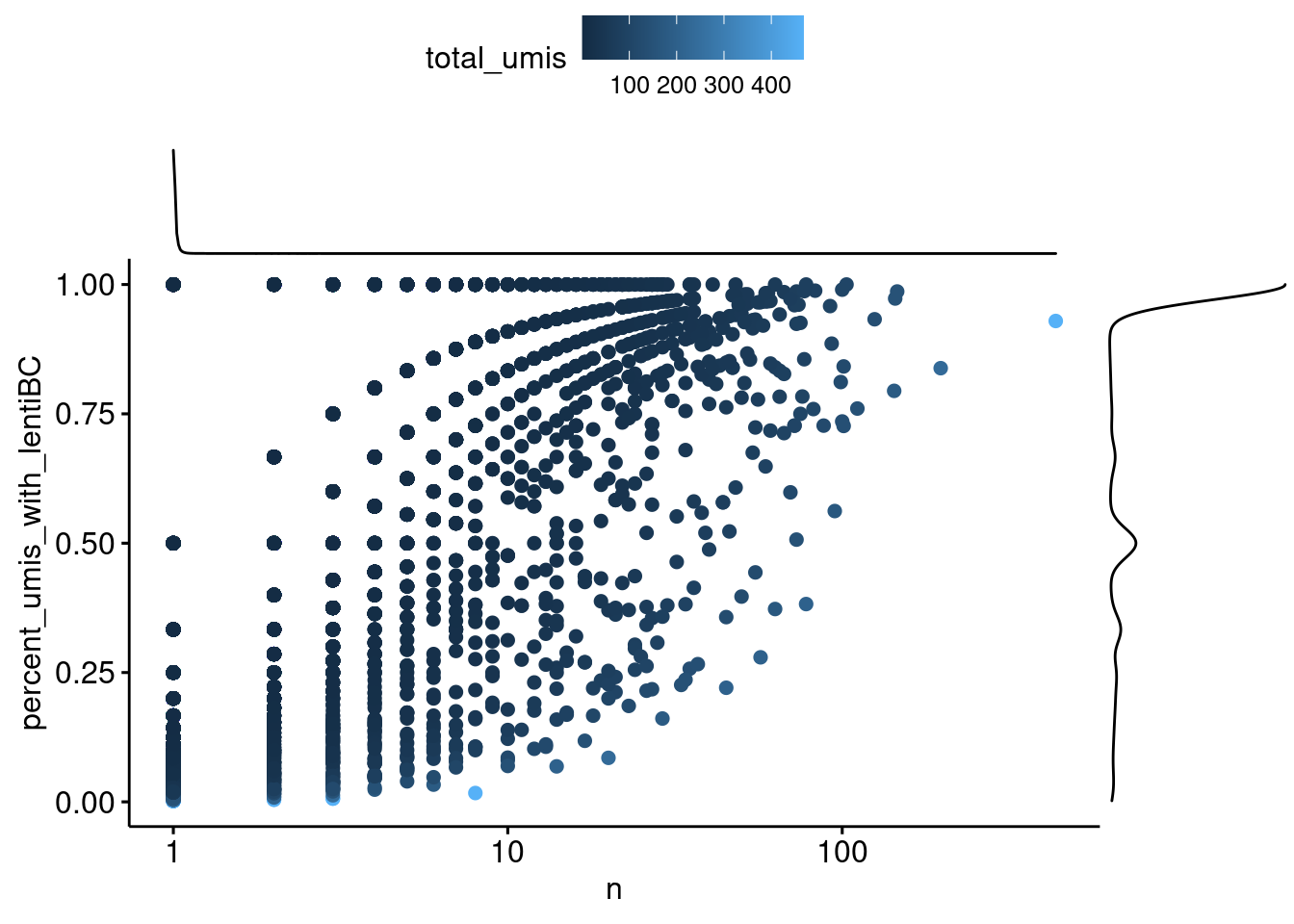
Capture4: Each point is a cell-lenti barcode pair. The x-axis is the number of umi supporting that pair. The y-axis is the percent of the total number of umi for the cell barcode that represents, where a 1 means that all umis from that cell have the same lenti barcode.
c5_barcode_umi_counts <- c5_both %>% distinct(cell_barcode, lenti_bc, umi) %>%
group_by(cell_barcode, lenti_bc) %>%
tally() %>% group_by(cell_barcode) %>%
mutate(percent_umis_with_lentiBC = n/sum(n),
total_umis = sum(n))
plot_pCells_c5 <- ggscatterhist(c5_barcode_umi_counts, x="n",
y="percent_umis_with_lentiBC",
color="total_umis", xscale = "log10") 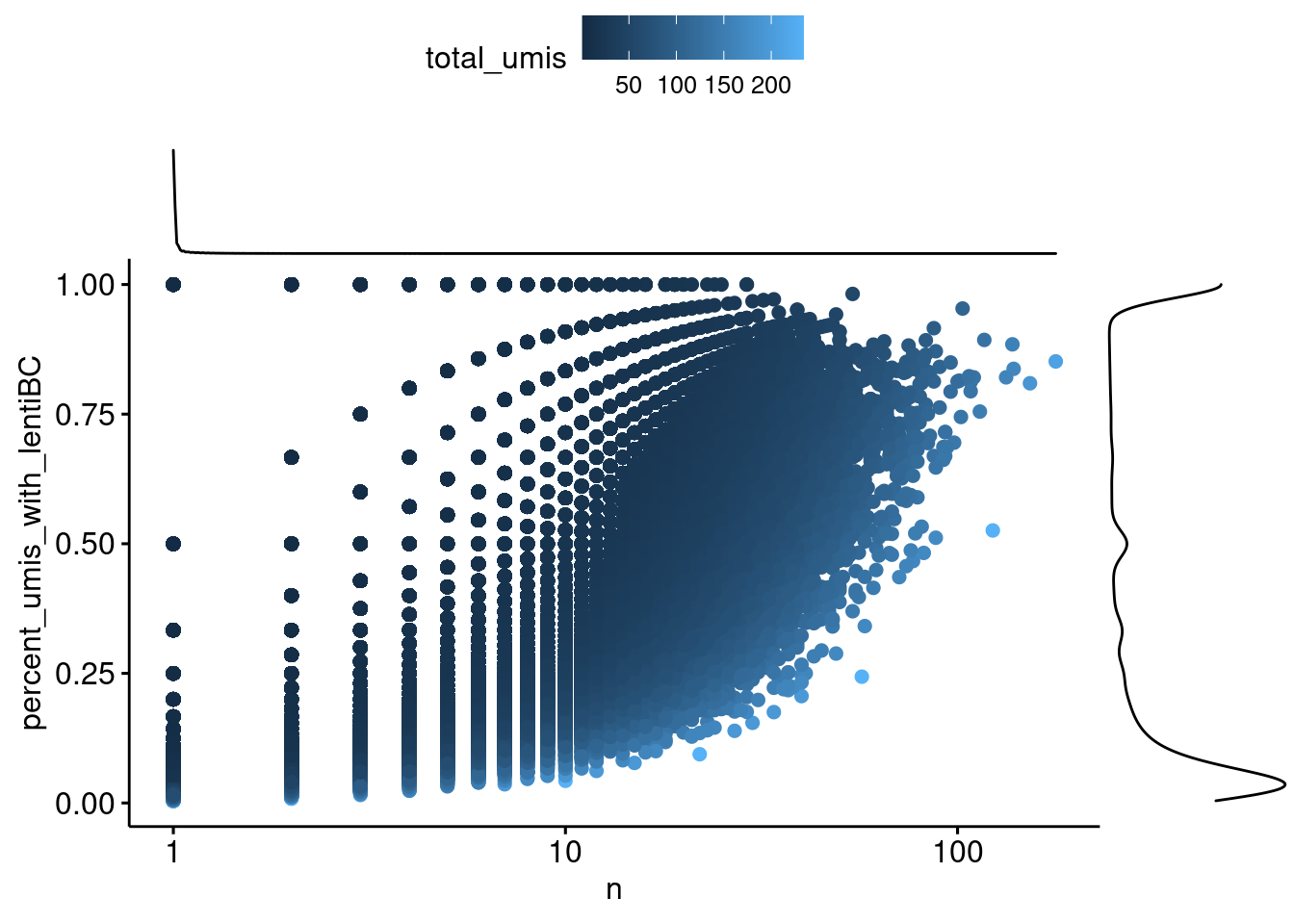
Capture5: Each point is a cell-lenti barcode pair. The x-axis is the number of umi supporting that pair. The y-axis is the percent of the total number of umi for the cell barcode that represents, where a 1 means that all umis from that cell have the same lenti barcode.
A summary of the number of cell barcodes meeting different criteria for inclusion:
c4_barcode_umi_counts_3 <- subset(c4_barcode_umi_counts, total_umis >= 3)
c5_barcode_umi_counts_3 <- subset(c5_barcode_umi_counts, total_umis >= 3)
lenti_cell_stats <- t(data.frame(list(
nCells = c(length(unique(c4_barcode_umi_counts$cell_barcode)),
length(unique(c5_barcode_umi_counts$cell_barcode))),
cells_with_1_lenti = c(nrow(subset(c4_barcode_umi_counts,
percent_umis_with_lentiBC == 1)),
nrow(subset(c5_barcode_umi_counts,
percent_umis_with_lentiBC == 1))),
nCells_3umi = c(length(unique(c4_barcode_umi_counts_3$cell_barcode)),
length(unique(c5_barcode_umi_counts_3$cell_barcode))),
umi3_cells_with_1_lenti = c(nrow(subset(c4_barcode_umi_counts_3,
percent_umis_with_lentiBC == 1)),
nrow(subset(c5_barcode_umi_counts_3,
percent_umis_with_lentiBC == 1))),
umi3_cells_with_80p_SameLenti = c(nrow(subset(c4_barcode_umi_counts_3,
percent_umis_with_lentiBC >= 2/3)),
nrow(subset(c5_barcode_umi_counts_3,
percent_umis_with_lentiBC >= 2/3))))))
colnames(lenti_cell_stats) <- c("Capture4", "Capture5")
lenti_cell_stats Capture4 Capture5
nCells 60256 194328
cells_with_1_lenti 50846 135378
nCells_3umi 10490 53053
umi3_cells_with_1_lenti 4510 2324
umi3_cells_with_80p_SameLenti 8173 12880We will keep cell barcodes that have support from at least 3 UMIs and where at least 66.6% of the UMIs are for the same lenti-barcode.
c4_good <- subset(c4_barcode_umi_counts_3, percent_umis_with_lentiBC >= 2/3)
c5_good <- subset(c5_barcode_umi_counts_3, percent_umis_with_lentiBC >= 2/3)
message("Number of capture 4 and capture 5 cells remaining: ",
nrow(c4_good), ", ", nrow(c5_good))Number of capture 4 and capture 5 cells remaining: 8173, 12880Generage lenti to cell barcode key for Captures 4 to 5
While each cell barcode should ideally only have 1 lenti barcode, a lenti barcode might be associated with many cell barcodes. Here is the distribution:
c4_good_con <- c4_good %>% group_by(lenti_bc) %>%
mutate(cell_barcodes = paste0(cell_barcode, collapse = ";")) %>%
mutate(n = str_count(cell_barcodes, ";") + 1) %>%
distinct(lenti_bc, .keep_all= TRUE) %>%
select(lenti_bc, n, cell_barcodes)
summary(c4_good_con$n) Min. 1st Qu. Median Mean 3rd Qu. Max.
1.00 1.00 2.00 10.93 5.00 3075.00 p4 <- ggplot(c4_good_con, aes(log10(n))) + geom_histogram(bins = 100) + theme_cowplot()
c5_good_con <- c5_good %>% group_by(lenti_bc) %>%
mutate(cell_barcodes = paste0(cell_barcode, collapse = ";")) %>%
mutate(n = str_count(cell_barcodes, ";") + 1) %>%
distinct(lenti_bc, .keep_all= TRUE) %>%
select(lenti_bc, n, cell_barcodes)
summary(c5_good_con$n) Min. 1st Qu. Median Mean 3rd Qu. Max.
1.00 1.00 2.00 14.89 6.00 6429.00 p5 <- ggplot(c5_good_con, aes(log10(n))) + geom_histogram(bins = 100) + theme_cowplot()
if(save){
write.table(c4_good_con, paste0(out, "Capture4-Lenti_cellbarcodes.tsv"),
sep = "\t", quote=FALSE, row.names = FALSE)
write.table(c5_good_con, paste0(out, "Capture5-Lenti_cellbarcodes.tsv"),
sep = "\t", quote=FALSE, row.names = FALSE)}
plot_grid(p4, p5)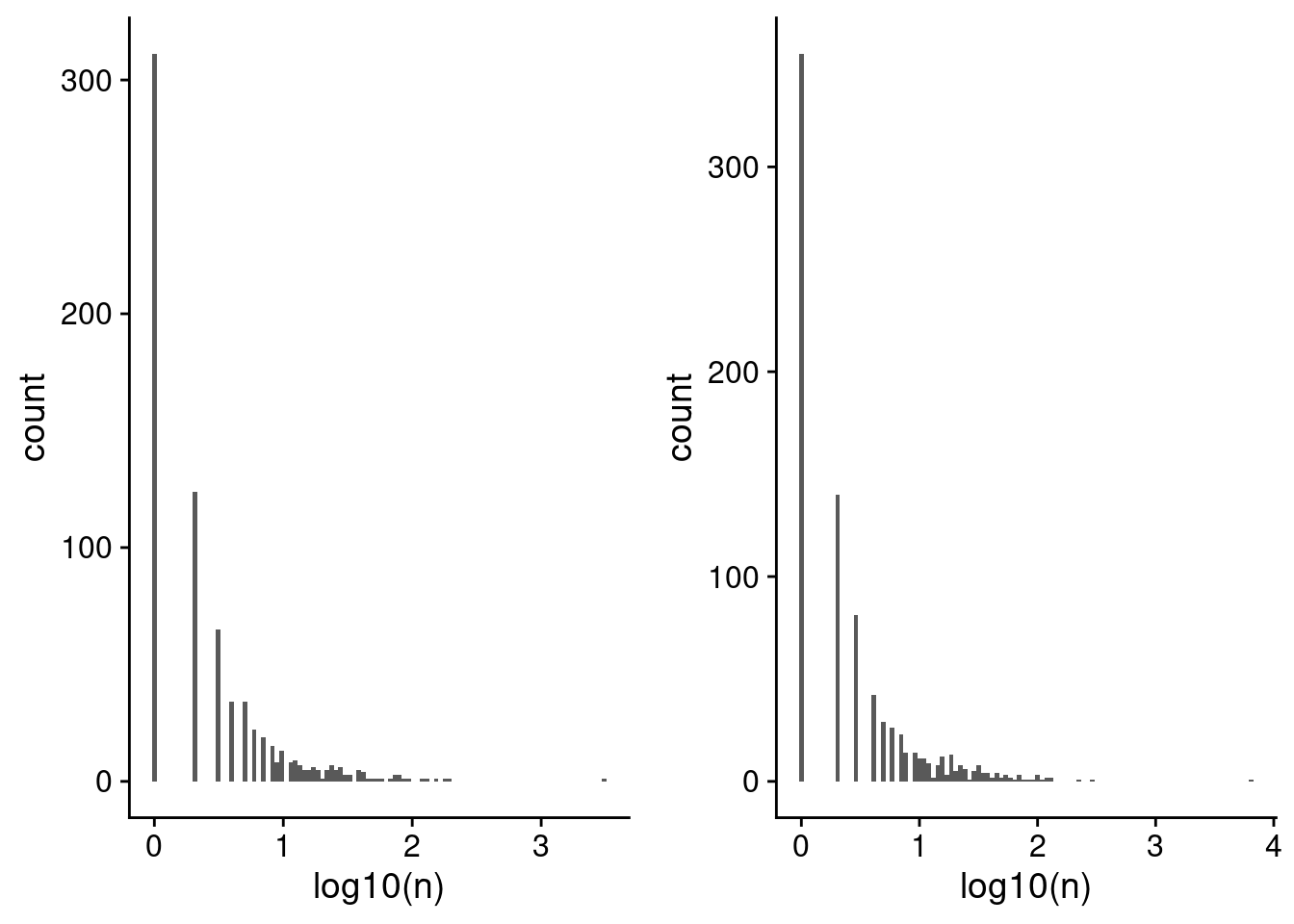
Distribution of the number of cell barcodes assigned to each lenti-barcode for (a) Capture 4 and (b) capture 5. Note the x-axis is on the log10 scale, as both have an extreme outlier.
Merge the two keys, this is the total number of lenti-barcodes that map between captures 4 and 5:
key <- c4_good_con %>% inner_join(c5_good_con, by = "lenti_bc", suffix = c(".Capture4", ".Capture5"))
nrow(key)[1] 571if(save){write.table(key, paste0(out, "Lenti2cell_barcode_key.tsv"), sep = "\t",
quote=FALSE, row.names = FALSE)}
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux 8.5 (Ootpa)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] stringr_1.4.0 viridis_0.6.2 viridisLite_0.4.0 cowplot_1.1.1
[5] ggpubr_0.4.0 ggplot2_3.3.5 data.table_1.14.2 tidyr_1.1.4
[9] dplyr_1.0.7 argparse_2.1.3
loaded via a namespace (and not attached):
[1] tidyselect_1.1.1 xfun_0.28 bslib_0.3.1 purrr_0.3.4
[5] carData_3.0-4 colorspace_2.0-2 vctrs_0.3.8 generics_0.1.1
[9] htmltools_0.5.2 yaml_2.2.1 utf8_1.2.2 rlang_0.4.12
[13] jquerylib_0.1.4 later_1.3.0 pillar_1.6.4 glue_1.6.0
[17] withr_2.4.3 DBI_1.1.1 lifecycle_1.0.1 munsell_0.5.0
[21] ggsignif_0.6.3 gtable_0.3.0 workflowr_1.6.2 evaluate_0.14
[25] labeling_0.4.2 knitr_1.36 fastmap_1.1.0 httpuv_1.6.5
[29] fansi_1.0.0 highr_0.9 broom_0.7.10 Rcpp_1.0.7
[33] backports_1.4.1 promises_1.2.0.1 scales_1.1.1 jsonlite_1.7.2
[37] abind_1.4-5 farver_2.1.0 fs_1.5.2 gridExtra_2.3
[41] digest_0.6.29 stringi_1.7.6 rstatix_0.7.0 rprojroot_2.0.2
[45] grid_4.1.1 tools_4.1.1 magrittr_2.0.1 sass_0.4.0
[49] tibble_3.1.6 car_3.0-12 crayon_1.4.2 whisker_0.4
[53] pkgconfig_2.0.3 ellipsis_0.3.2 assertthat_0.2.1 rmarkdown_2.11
[57] R6_2.5.1 git2r_0.29.0 compiler_4.1.1