Pilot3: dropkick analysis
Christina Azodi
2022-03-01
Last updated: 2022-03-01
Checks: 5 2
Knit directory: BAUH_2020_MND-single-cell/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3.0_iPSC/01_cellcalling-dropkick/ | ../output/pilot3.0_iPSC/01_cellcalling-dropkick |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3.0_MN/01_cellcalling-dropkick/ | ../output/pilot3.0_MN/01_cellcalling-dropkick |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3f19b81. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .cache/
Ignored: .config/
Ignored: .nv/
Ignored: .snakemake/
Ignored: BAUH_2020_MND-single-cell.Rproj
Ignored: GRCh38_turboGFP-RFP_reference/
Ignored: Homo_sapiens.GRCh38.turboGFP/
Ignored: Rplots.pdf
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/pilot3_aggr-experiments.csv
Ignored: data/pilot3_donors.txt
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/2021-04-27_pilot2_nCells-per-donor.pdf
Ignored: output/2021-08-03_pilot2_nCells-per-donor.pdf
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: output/pilot2_testing/
Ignored: output/pilot3.0/
Ignored: output/pilot3.0_MN/
Ignored: output/pilot3.0_captures-separate/
Ignored: output/pilot3.0_iPSC/
Ignored: output/pilot3_Lenti/
Ignored: references/Homo_sapiens.GRCh38.turboGFP.bed
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa.fai
Ignored: references/Homo_sapiens.GRCh38.turboGFP.filtered.gtf
Ignored: references/Homo_sapiens.GRCh38.turboGFP.gtf
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Ignored: references/pilot3/
Ignored: references/test
Ignored: references/turboGFP.fa
Ignored: references/turboGFP.gtf
Ignored: references/turboRFP.fa
Ignored: workflow/rules/
Untracked files:
Untracked: Capture5-GEX/
Untracked: __Capture5-GEX.mro
Untracked: cellbender.dockerfile
Untracked: hwe1e-05_maf05_vcf_stats.txt
Untracked: hwe1e-05_vcf_stats.txt
Unstaged changes:
Modified: analysis/2022-03-01_pilot3_dropkick.Rmd
Modified: config/config_pilot3.0_MN.yml
Modified: config/config_pilot3.0_iPSC.yml
Modified: workflow/Snakefile
Modified: workflow/rules_cellcalling.smk
Modified: workflow/rules_demultiplexing.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/2022-03-01_pilot3_dropkick.Rmd) and HTML (public/2022-03-01_pilot3_dropkick.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 9eb9fcc | cazodi | 2022-03-01 | add rmd for dropkick and dropletqc |
| html | 9eb9fcc | cazodi | 2022-03-01 | add rmd for dropkick and dropletqc |
dropkick cellcalling
Dropkick is a fully automated software for QC and filtering scRNA-seq data with a focus on excluding ambient barcodes and recovering only real cells. By assigning training labels based on predictive global heuristics, dropkick learns a gene-based representation of real cells and ambient noise, calculating a cell probability score for each barcode (where a higher score means more likly a cell). Unlike EmptyDroplet, it does not set heuristic minimum thresholds for counts per cell.
Example results from the dropkick manuscript
Here is a QC summary and the results from applying dropkick to a pan T-cell dataset. Ideally, you want few genes with low dropout (if the gene is present in barcodes, it is very likely ambient):
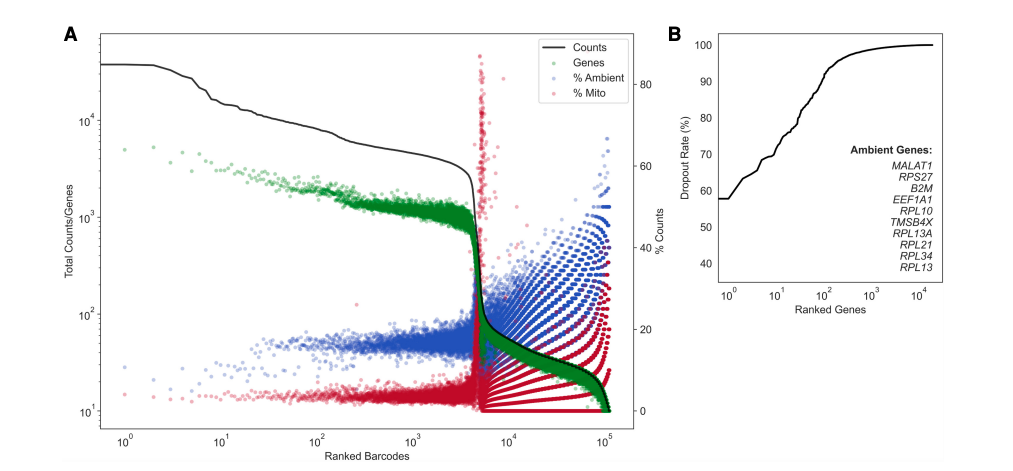
(left) a profile of total counts (black trace) and genes (green points) detected per ranked barcode, with the percentage of mitochondrial (red) and ambient (blue) reads for each barcode included to denote quality along dataset profile. (right) The dropout rate per gene ranked where top ranked genes are present in all barcodes (i.e. likely ambient). Ambient genes are identified by dropkick and used to calculate ambient percentage in the left figure.
For the cell probability score you want to see a clear separation between barcodes with a low (ambient) vs high (cell) score.
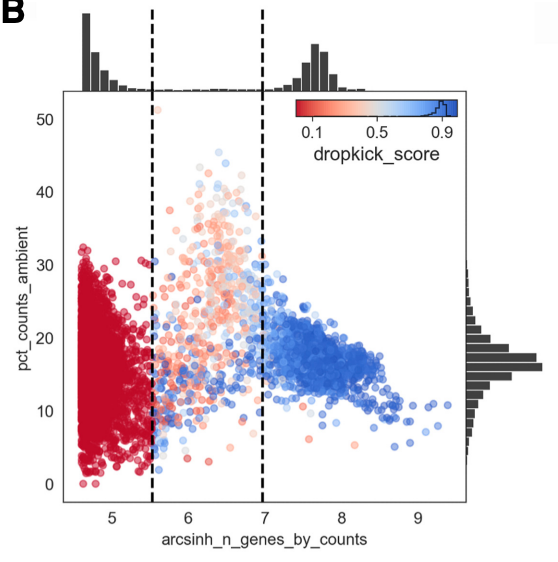
The percent ambient counts versus arcsinh-transformed genes detected per barcode, with histogram distributions plotted on margins. Initial dropkick thresholds defining the training set are shown as dashed vertical lines. Each point (barcode) is colored by its final dropkick score after model fitting.
Reminder: pilot 2 results
In our pilot 2 study, there were over 1,000 genes had a dropout rate near zero! Indicating very high ambient read levels.
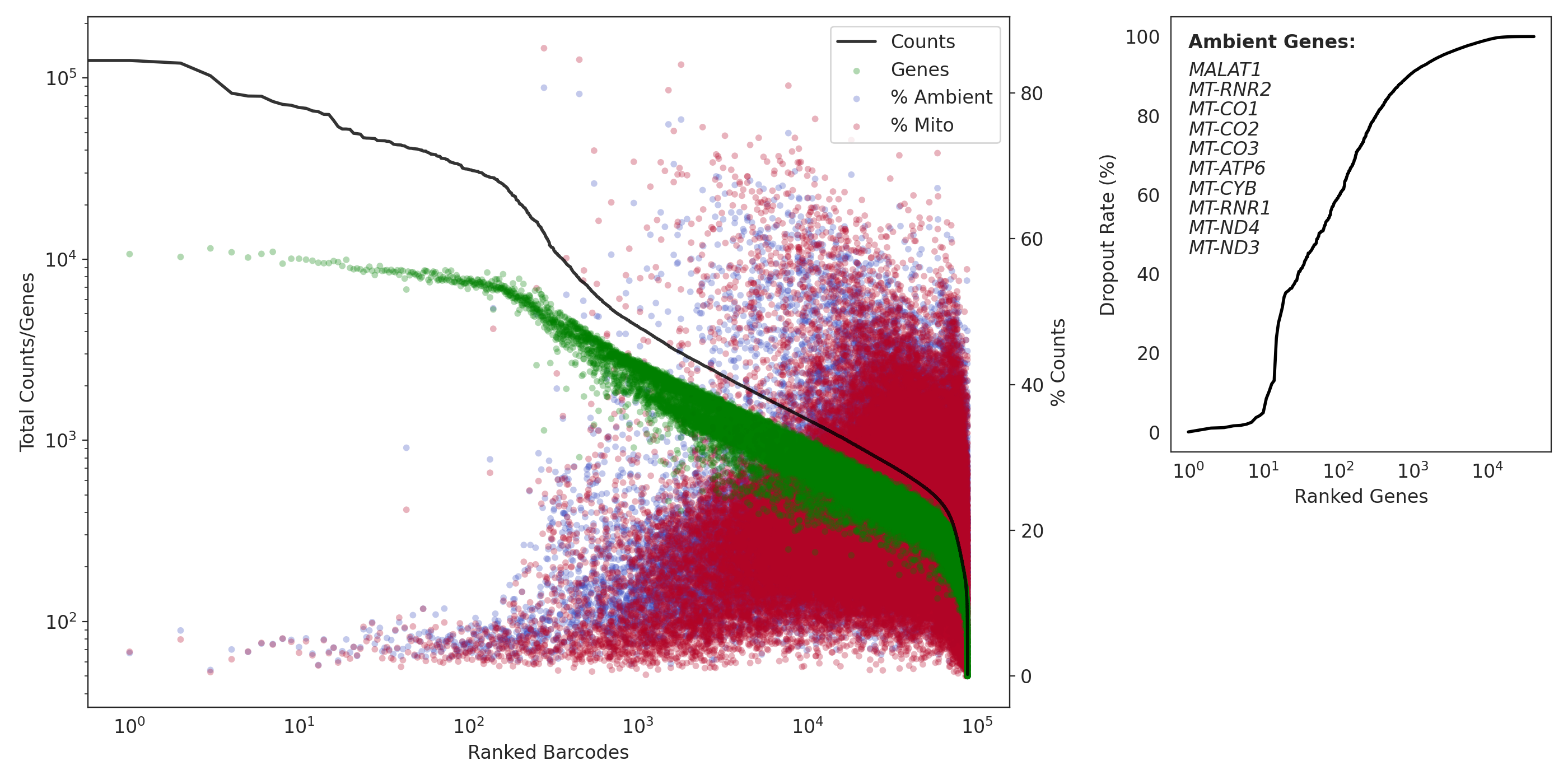
(QC: pilot 2)
Dropkick also had a difficult time separating ambient from cells.
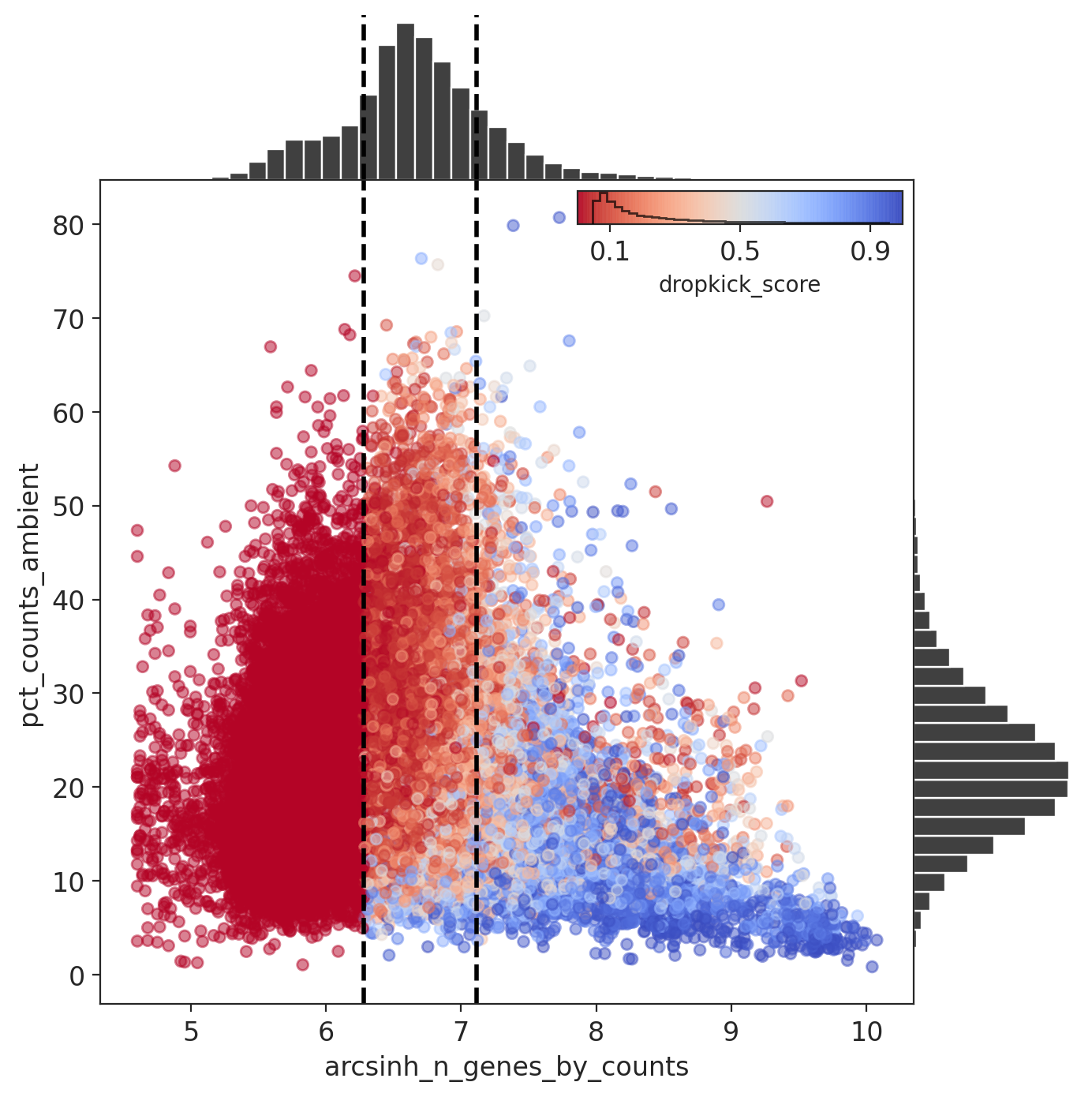
Results: pilot2
iPSCs (Capture 5)
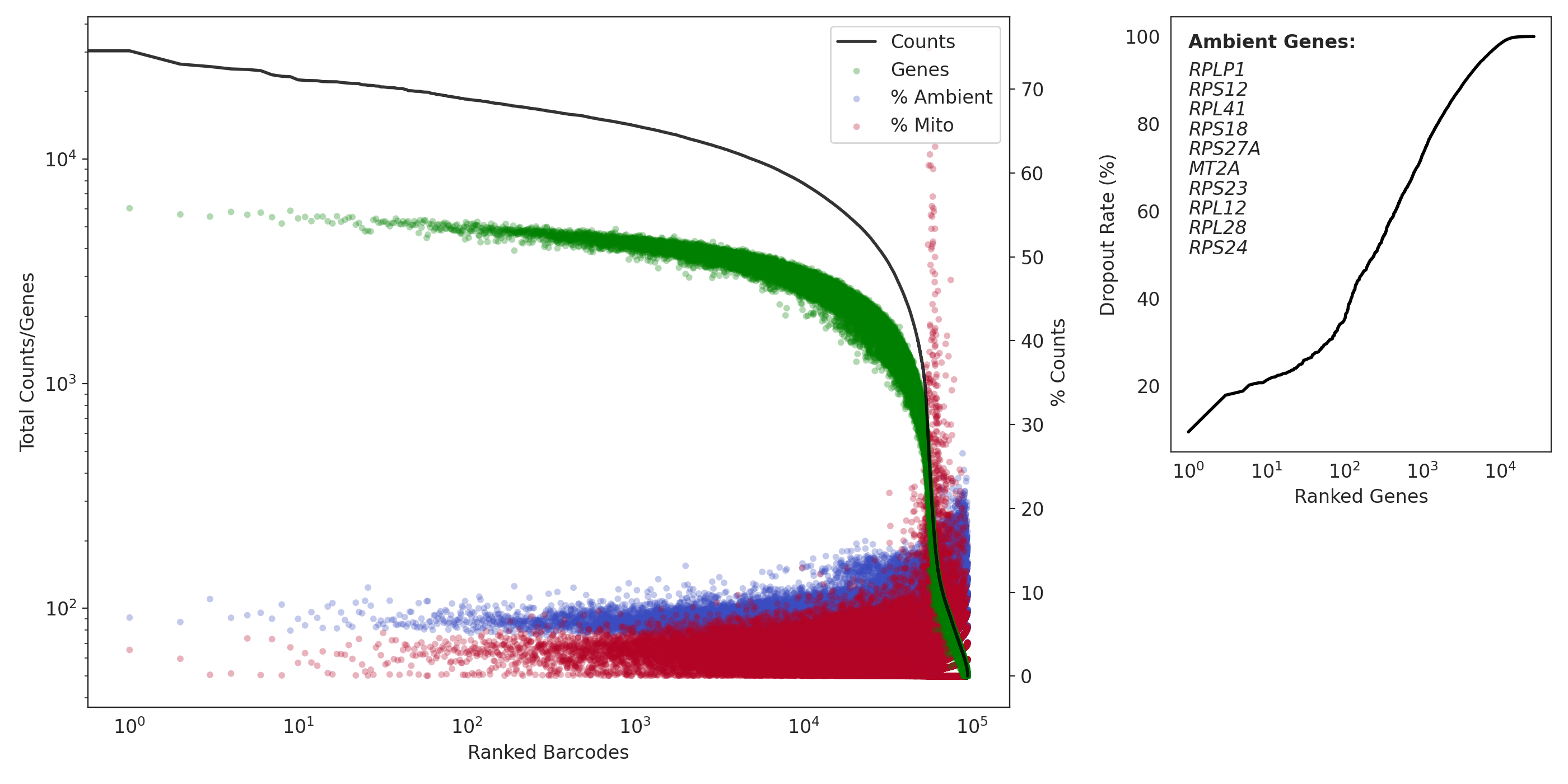
QC: Capture 5 (iPSCs)
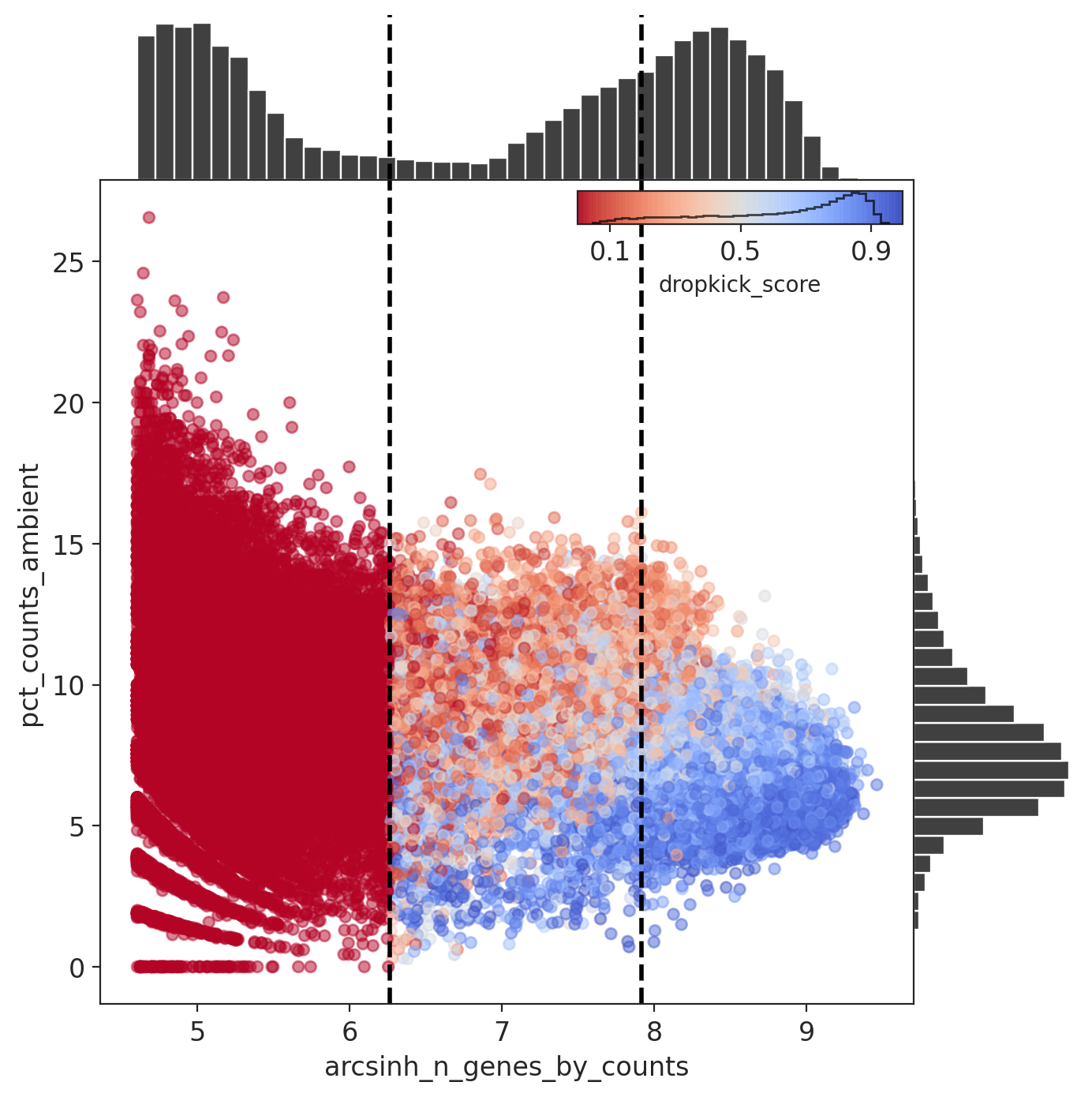
Results: Capture 5 (iPSCs)
Summary: Clear separation between barcodes with high and low ratio of genes to counts, with many confident cell calls, but still a wide tail, suggesting many barcodes are difficult to classify.
Number of cells called:
bc_c5 <- scan(paste0(ipsc_dir, "Capture5-GEX/raw_feature_bc_matrix_dropkick_barcodes.txt"),
what="character")
message("Capture 5: ", length(bc_c5))Capture 5: 40466MNs (Captures 1-4)
QC Plots
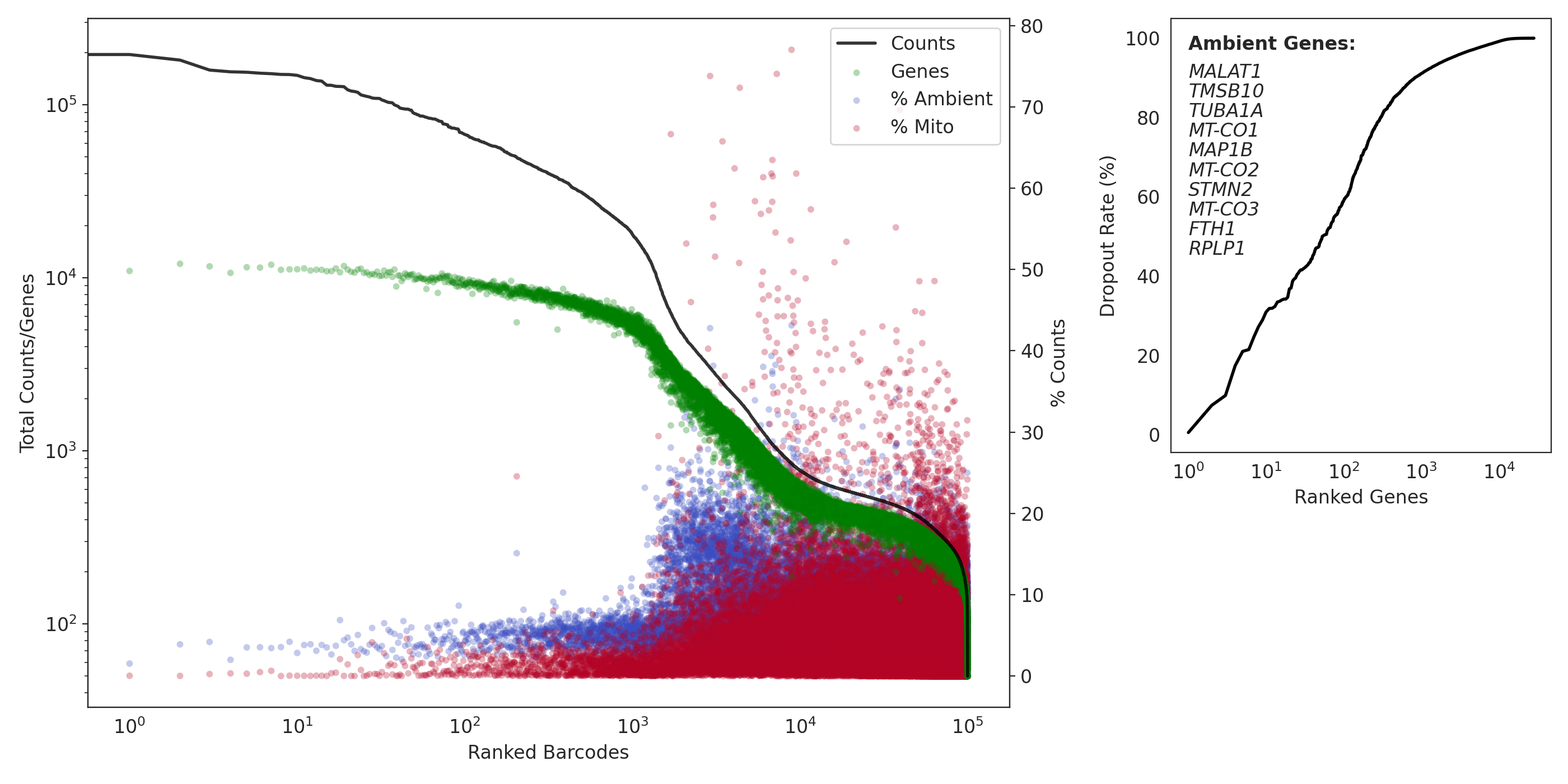
QC: Capture 1 (MNs)
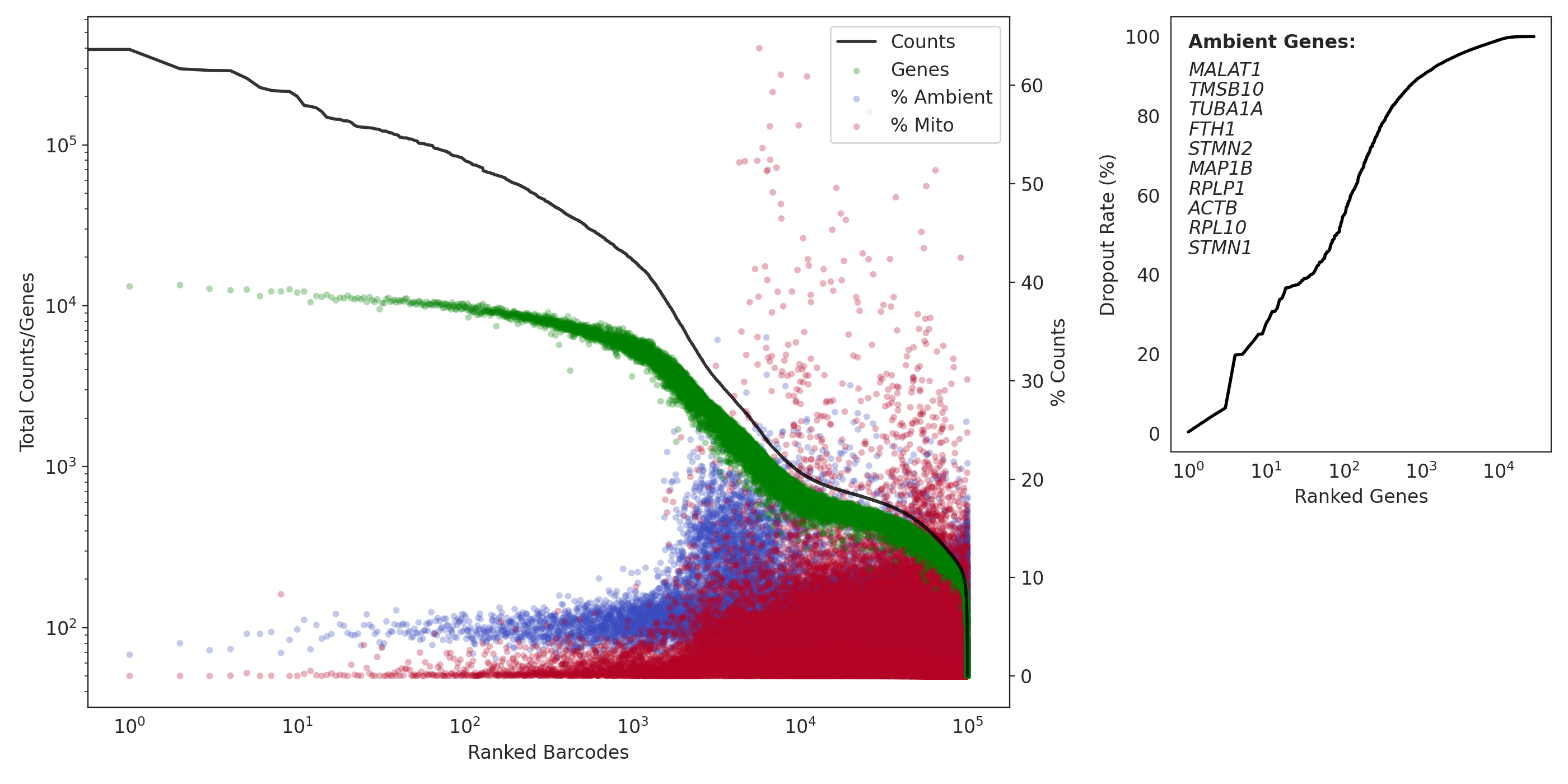
QC: Capture 2 (MNs)
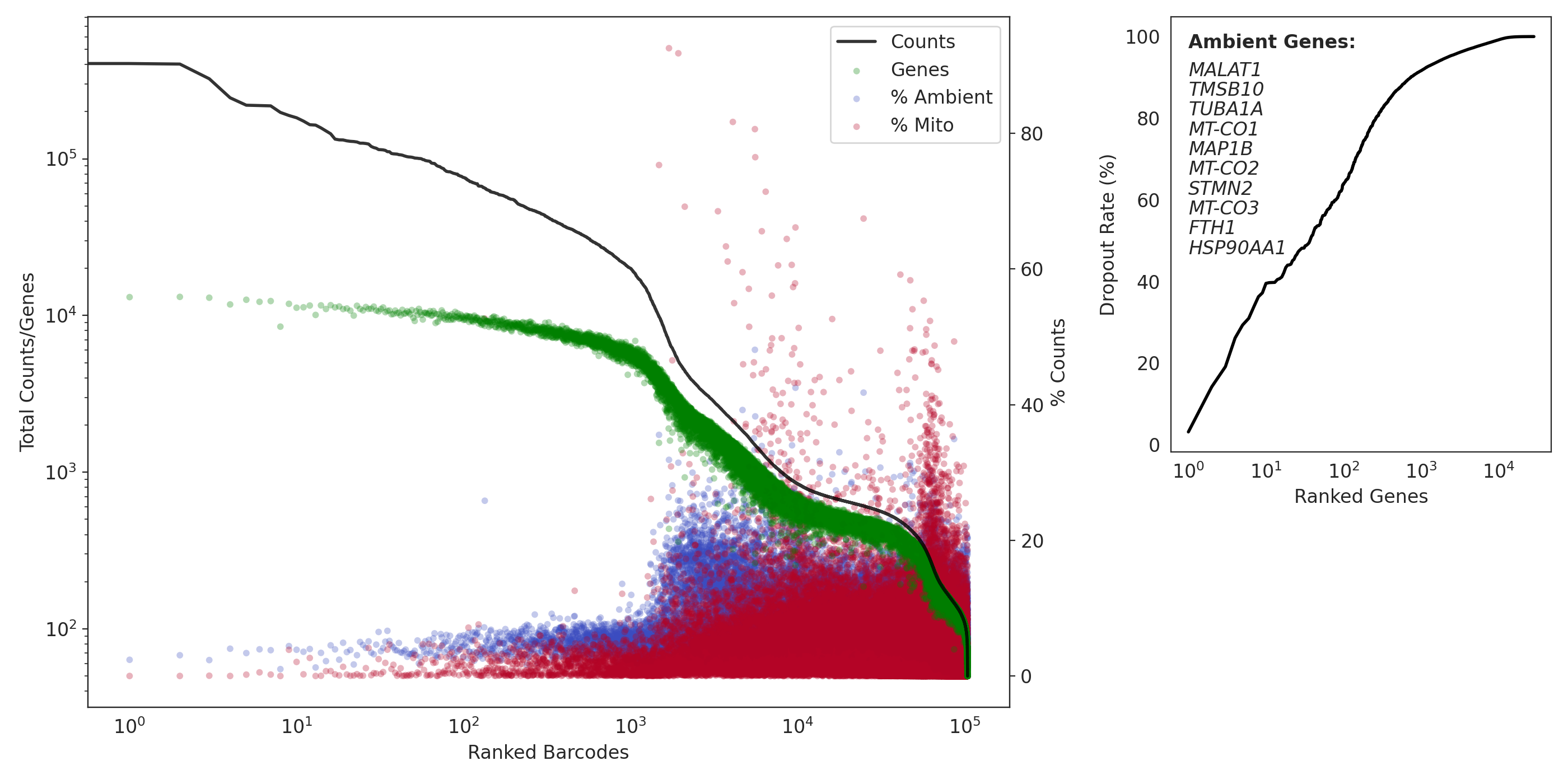
QC: Capture 3 (MNs)
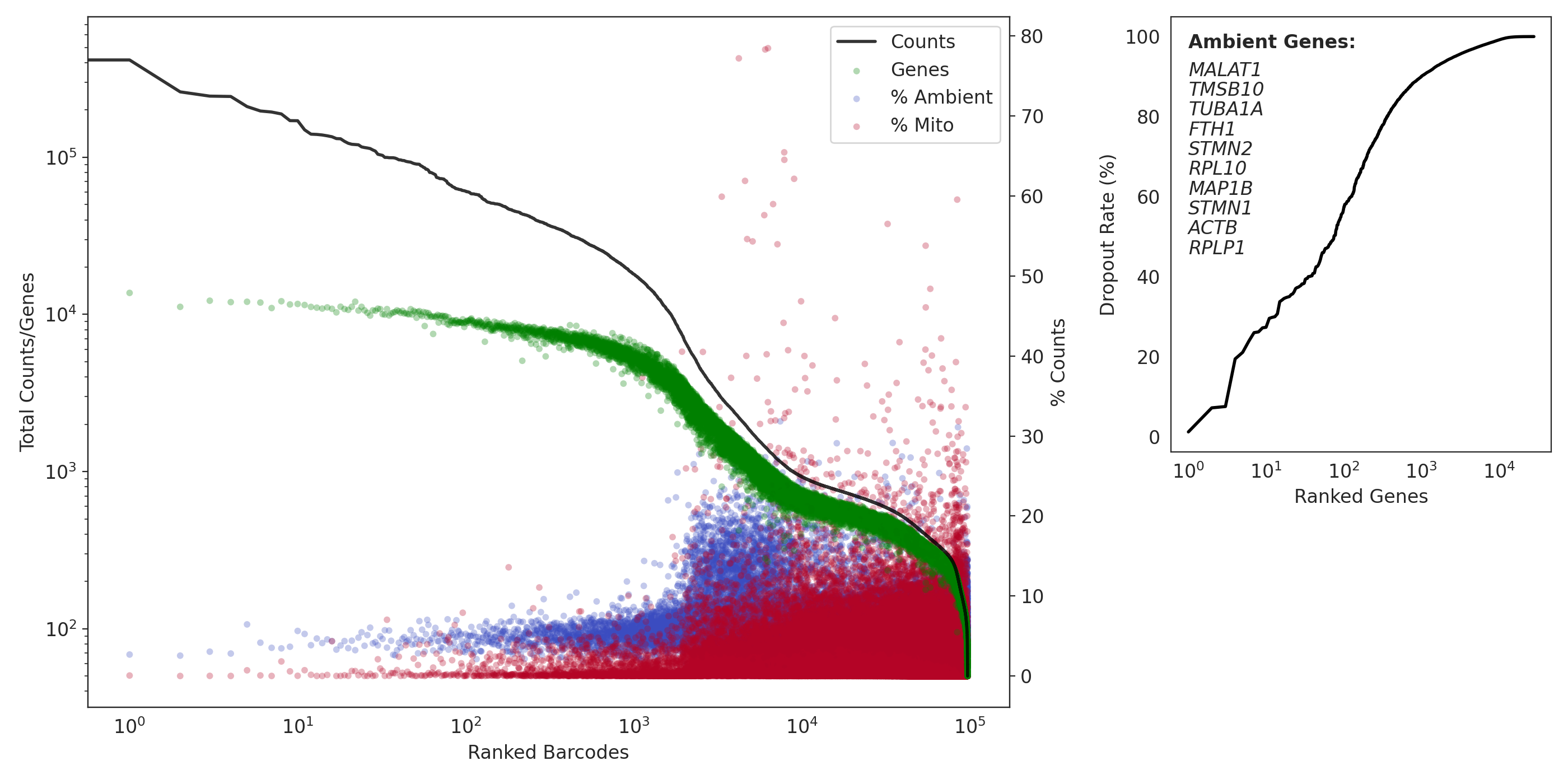
QC: Capture 4 (MNs)
Results Plots
In all four MN captures, most barcodes were given a low dropkick score (<0.2).
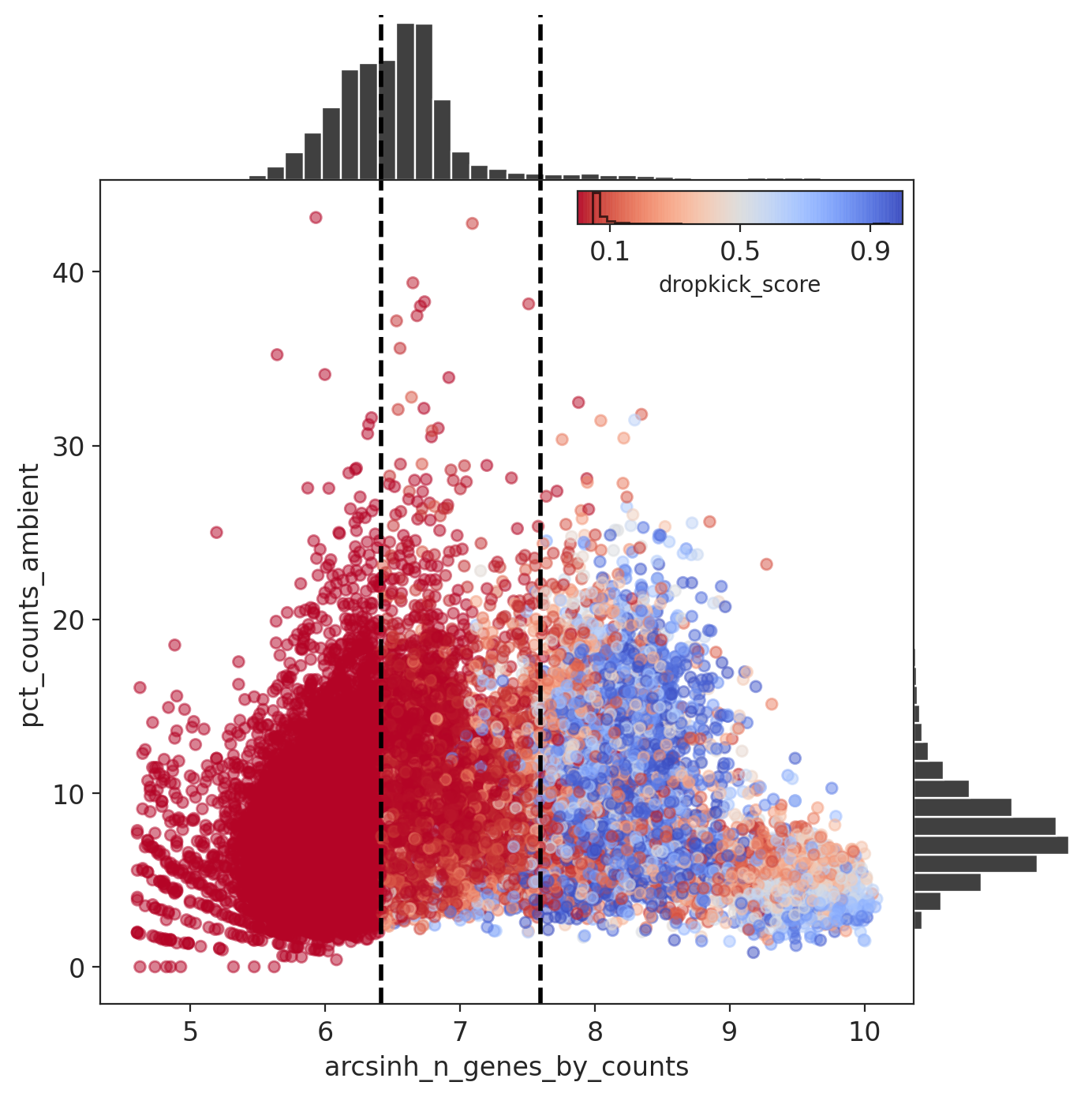
Results: Capture 1 (MNs)
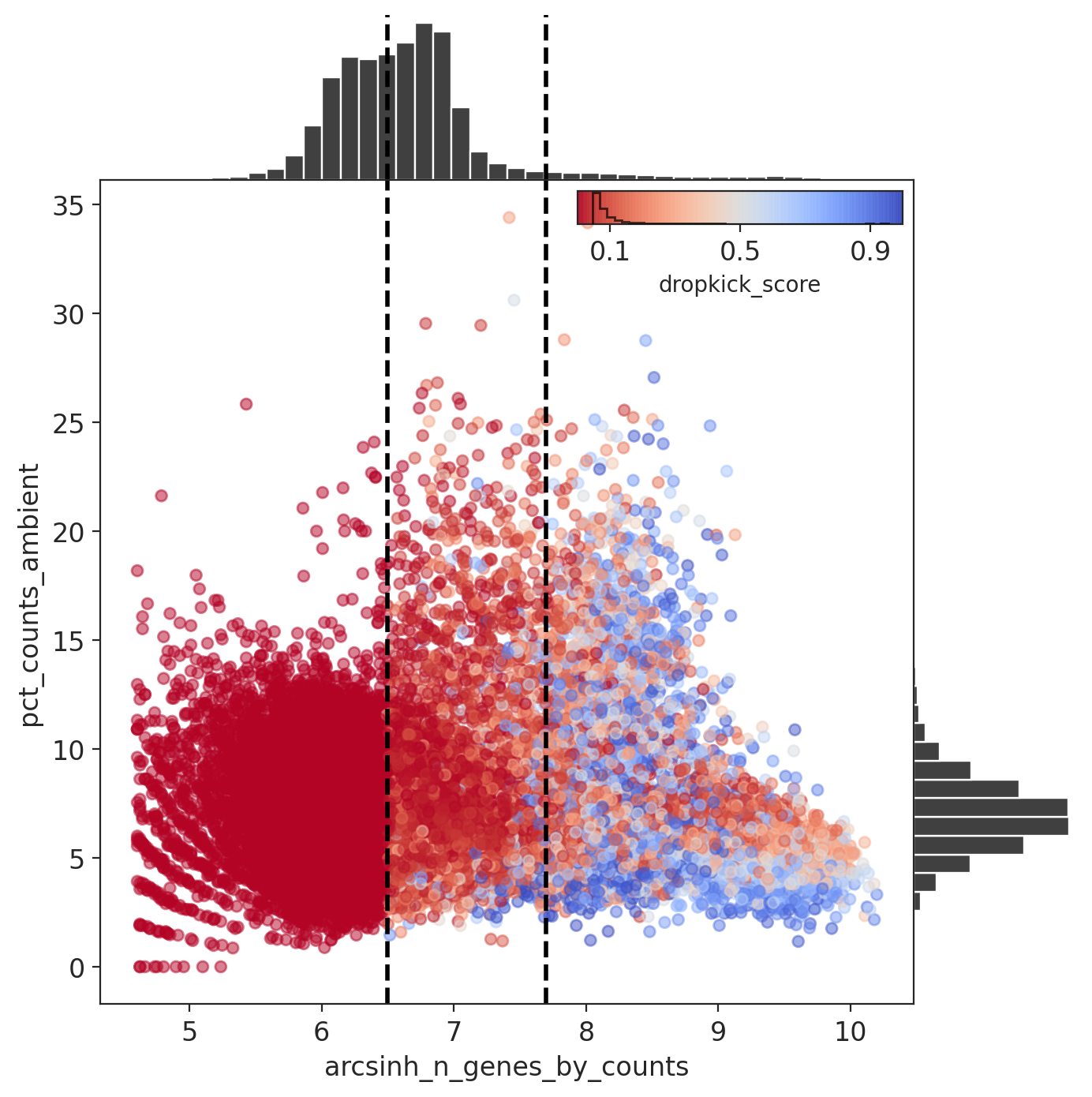
Results: Capture 2 (MNs)
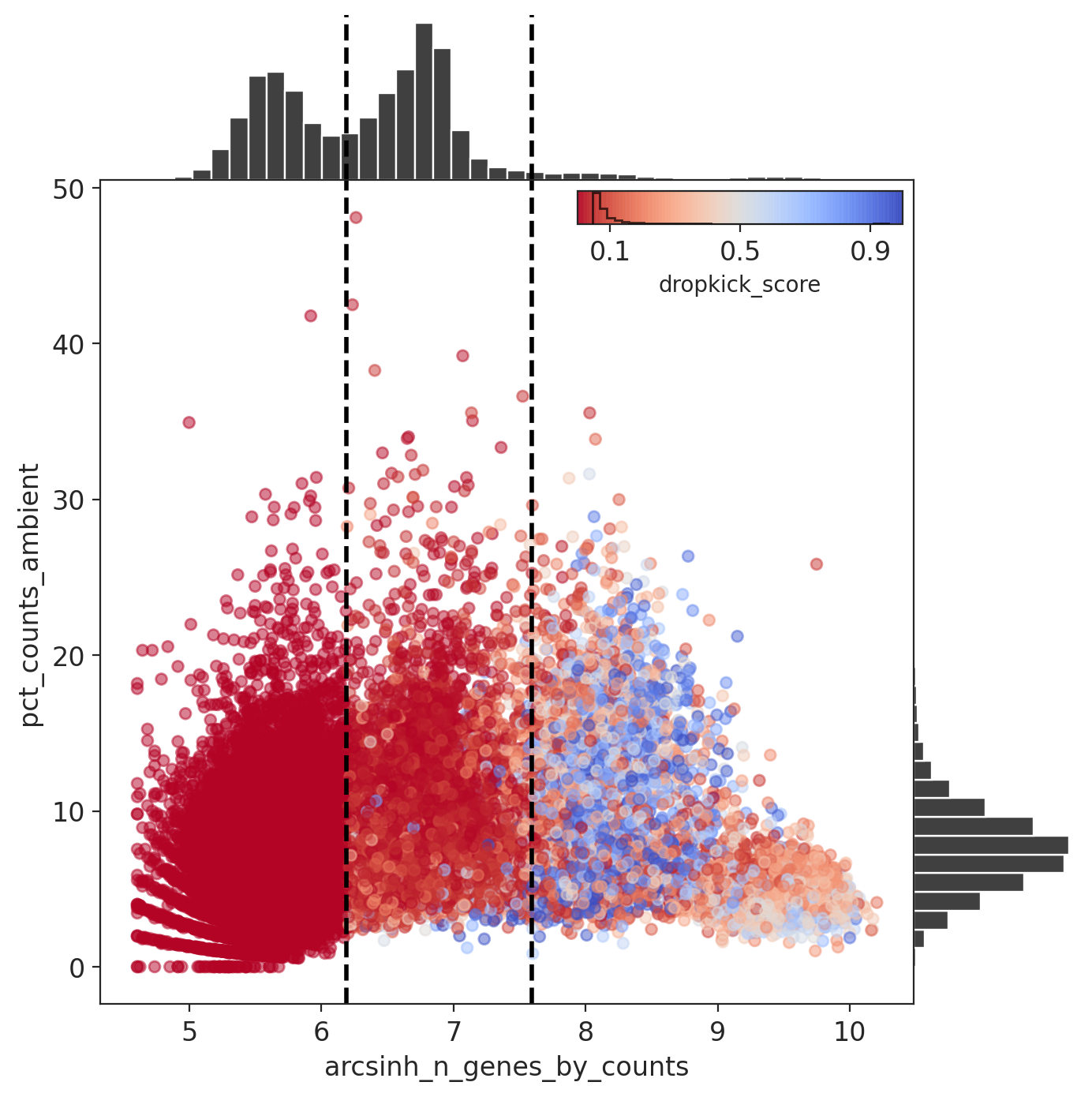
Results: Capture 3 (MNs)
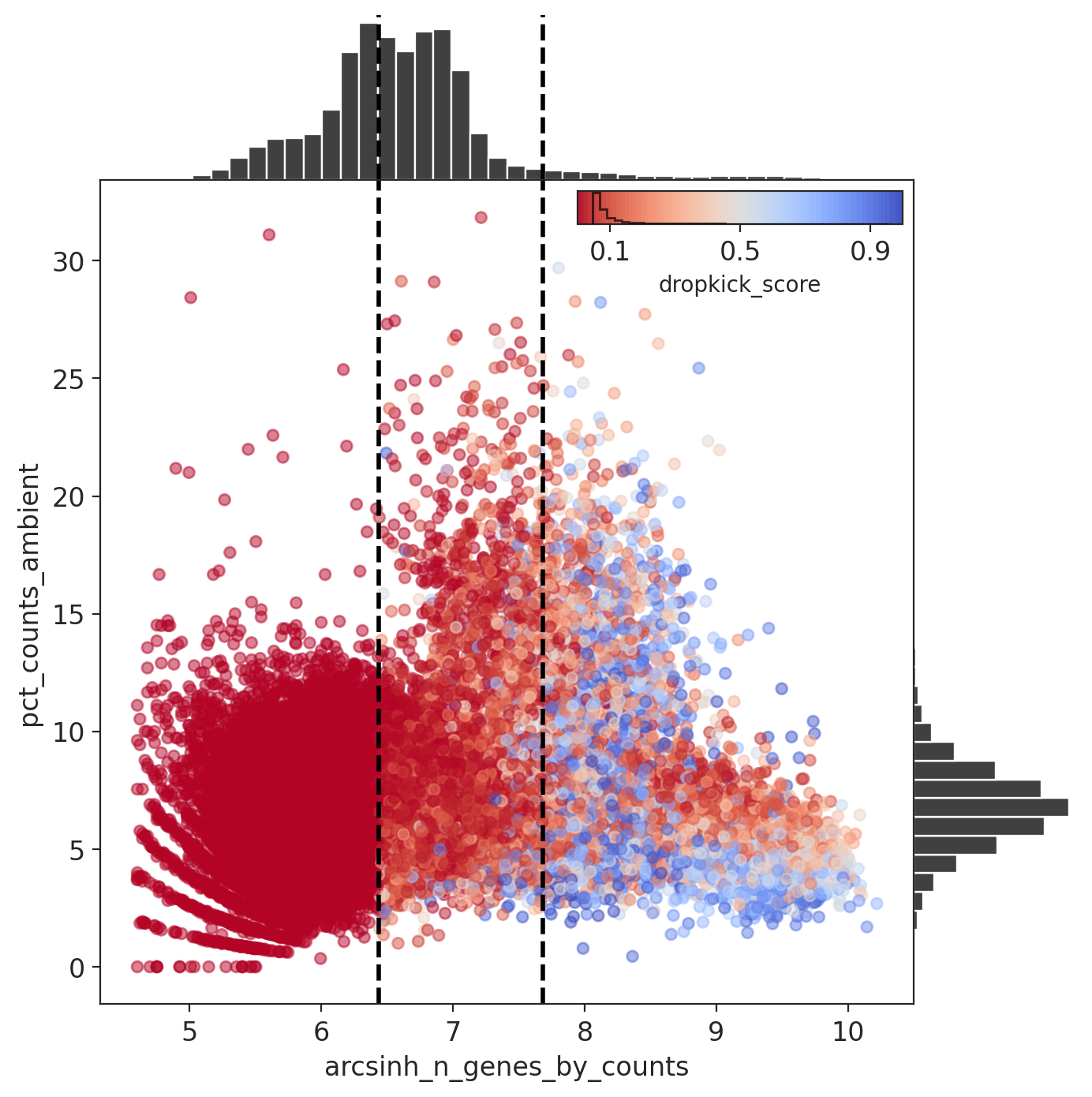
Results: Capture 4 (MNs)
bc_c1 <- scan(paste0(mn_dir, "Capture1-GEX/raw_feature_bc_matrix_dropkick_barcodes.txt"),
what="character")
message("Capture 1: ", length(bc_c1))Capture 1: 3586bc_c2 <- scan(paste0(mn_dir, "Capture2-GEX/raw_feature_bc_matrix_dropkick_barcodes.txt"),
what="character")
message("Capture 2: ", length(bc_c2))Capture 2: 3207bc_c3 <- scan(paste0(mn_dir, "Capture3-GEX/raw_feature_bc_matrix_dropkick_barcodes.txt"),
what="character")
message("Capture 3: ", length(bc_c3))Capture 3: 2960bc_c4 <- scan(paste0(mn_dir, "Capture4-GEX/raw_feature_bc_matrix_dropkick_barcodes.txt"),
what="character")
message("Capture 4: ", length(bc_c4))Capture 4: 2793Compare ambient genes
amb1 <- fread(paste0(mn_dir, "Capture1-GEX/raw_feature_bc_matrix_dropkick_ambient-features.txt"),
sep=",")
amb2 <- fread(paste0(mn_dir, "Capture2-GEX/raw_feature_bc_matrix_dropkick_ambient-features.txt"),
sep=",")
amb3 <- fread(paste0(mn_dir, "Capture3-GEX/raw_feature_bc_matrix_dropkick_ambient-features.txt"),
sep=",")
amb4 <- fread(paste0(mn_dir, "Capture4-GEX/raw_feature_bc_matrix_dropkick_ambient-features.txt"),
sep=",")
amb5 <- fread(paste0(ipsc_dir, "Capture5-GEX/raw_feature_bc_matrix_dropkick_ambient-features.txt"),
sep=",")
geneList <- list(Capture1 = amb1$gene_ids,
Capture2 = amb2$gene_ids,
Capture3 = amb3$gene_ids,
Capture4 = amb4$gene_ids)
ggvenn(geneList, stroke_size = 0.5, set_name_size = 4, text_size = 3,
fill_color = c("#0073C2FF", "#EFC000FF", "#868686FF", "#CD534CFF")) 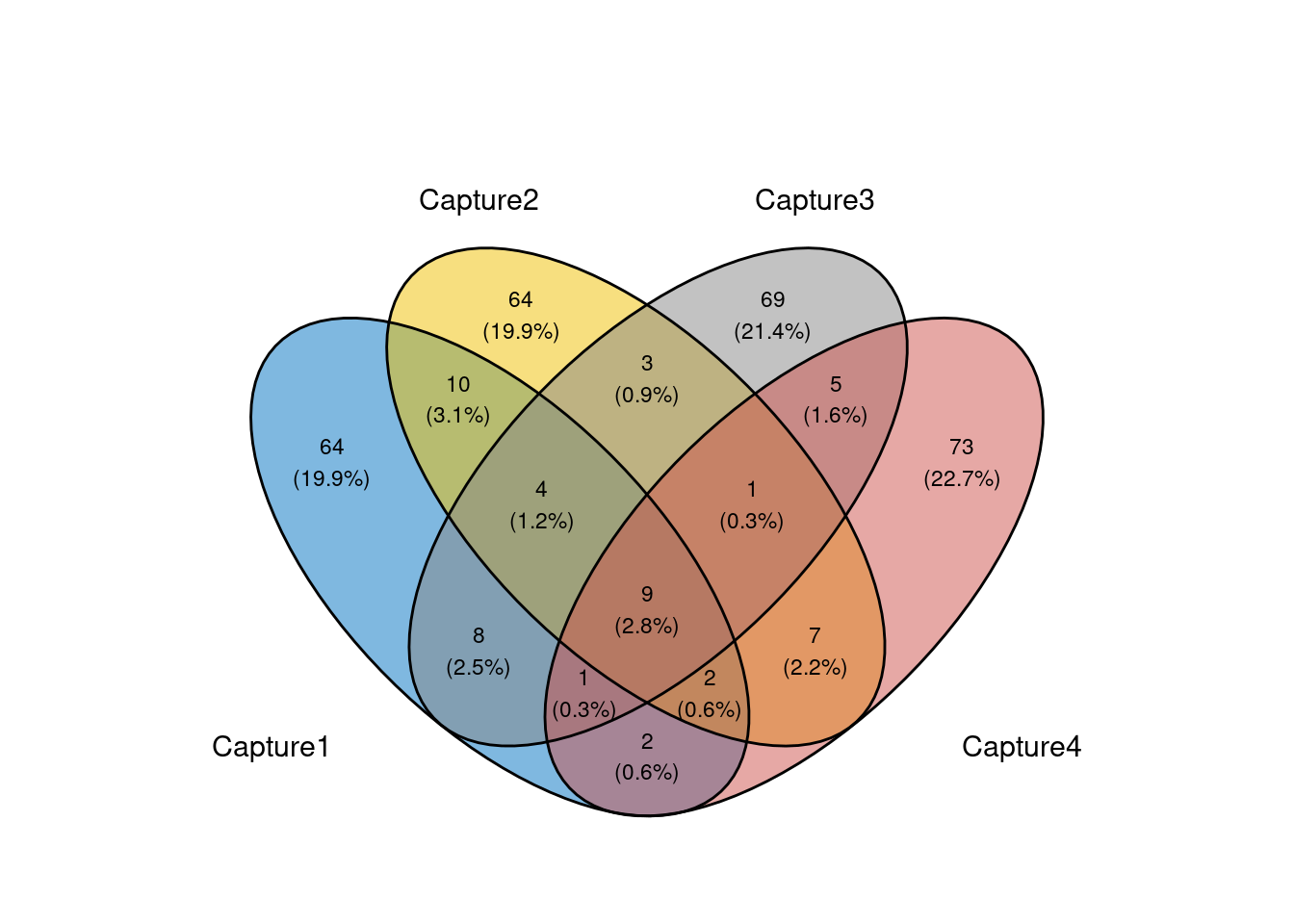
Venn diagram of overlap in top 100 ambient genes for each MN capture.
| Version | Author | Date |
|---|---|---|
| 9eb9fcc | cazodi | 2022-03-01 |
geneList2 <- list(MN_Captures = unique(c(amb1$gene_ids, amb2$gene_ids,
amb3$gene_ids, amb4$gene_ids)),
Capture5 = amb5$gene_ids)
ggvenn(geneList2, stroke_size = 0.5, set_name_size = 4, text_size = 3,
fill_color = c("#0073C2FF", "#EFC000FF", "#868686FF", "#CD534CFF")) 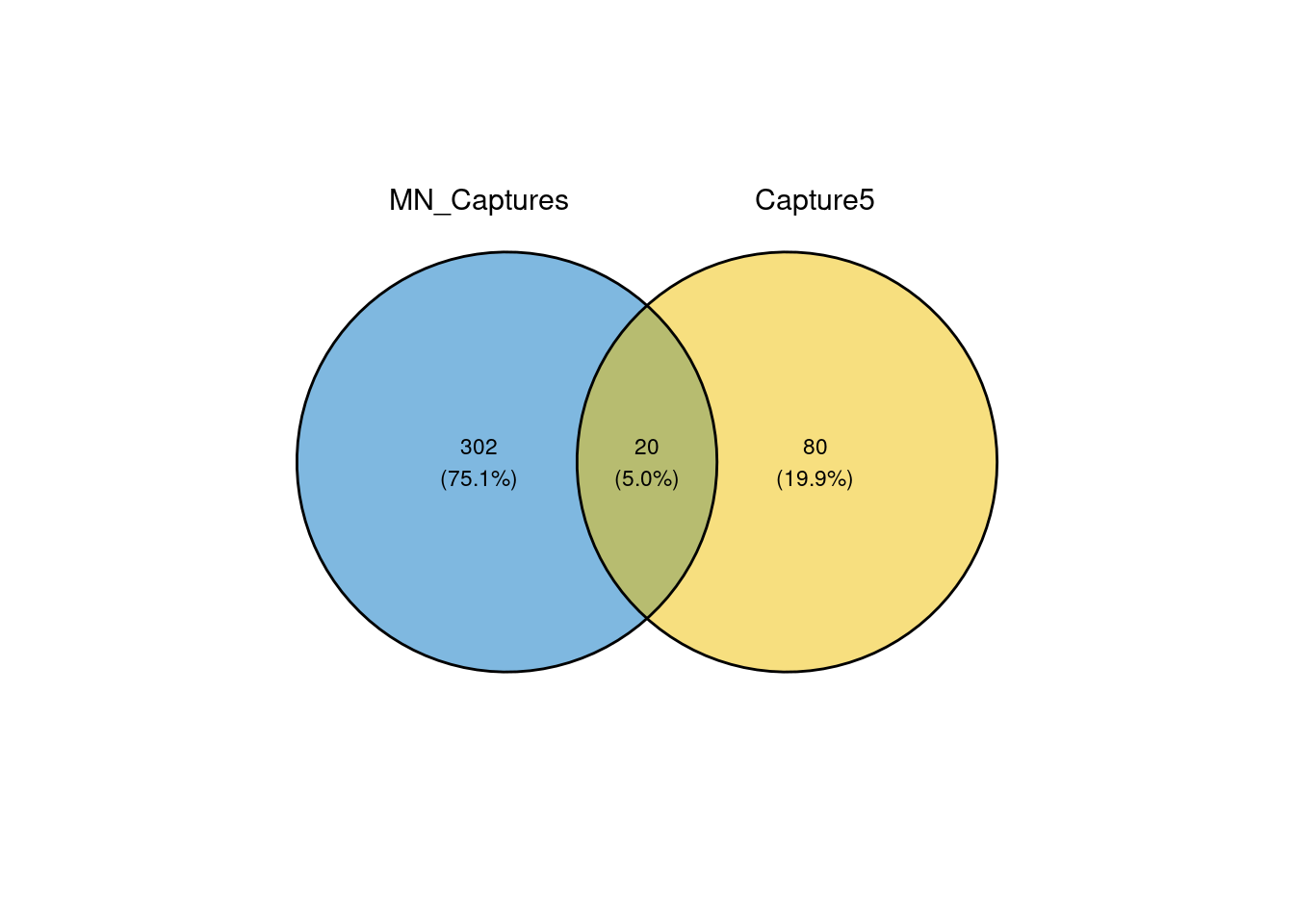
Venn diagram of overlap in top 100 ambient genes from iPSCs with top 100 ambient genes from all 4 MN captures combined.
| Version | Author | Date |
|---|---|---|
| 9eb9fcc | cazodi | 2022-03-01 |
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux 8.5 (Ootpa)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ComplexHeatmap_2.10.0 ggvenn_0.1.9 ggplot2_3.3.5
[4] data.table_1.14.2 tidyr_1.1.4 dplyr_1.0.7
[7] argparse_2.1.3
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 circlize_0.4.13 png_0.1-7
[4] assertthat_0.2.1 rprojroot_2.0.2 digest_0.6.29
[7] foreach_1.5.1 utf8_1.2.2 R6_2.5.1
[10] stats4_4.1.1 evaluate_0.14 highr_0.9
[13] pillar_1.6.4 GlobalOptions_0.1.2 rlang_0.4.12
[16] whisker_0.4 jquerylib_0.1.4 S4Vectors_0.32.3
[19] GetoptLong_1.0.5 rmarkdown_2.11 labeling_0.4.2
[22] stringr_1.4.0 munsell_0.5.0 compiler_4.1.1
[25] httpuv_1.6.5 xfun_0.28 pkgconfig_2.0.3
[28] BiocGenerics_0.40.0 shape_1.4.6 htmltools_0.5.2
[31] tidyselect_1.1.1 tibble_3.1.6 workflowr_1.6.2
[34] IRanges_2.28.0 codetools_0.2-18 matrixStats_0.61.0
[37] fansi_1.0.0 crayon_1.4.2 withr_2.4.3
[40] later_1.3.0 jsonlite_1.7.2 gtable_0.3.0
[43] lifecycle_1.0.1 DBI_1.1.1 git2r_0.29.0
[46] magrittr_2.0.1 scales_1.1.1 stringi_1.7.6
[49] farver_2.1.0 fs_1.5.2 promises_1.2.0.1
[52] doParallel_1.0.16 bslib_0.3.1 ellipsis_0.3.2
[55] generics_0.1.1 vctrs_0.3.8 rjson_0.2.20
[58] RColorBrewer_1.1-2 iterators_1.0.13 tools_4.1.1
[61] glue_1.6.0 purrr_0.3.4 parallel_4.1.1
[64] fastmap_1.1.0 yaml_2.2.1 clue_0.3-60
[67] colorspace_2.0-2 cluster_2.1.2 knitr_1.36
[70] sass_0.4.0