Pilot3: dropletQC analysis
Christina Azodi
2022-03-01
Last updated: 2022-03-01
Checks: 5 2
Knit directory: BAUH_2020_MND-single-cell/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3.0_iPSC/01_cellcalling-dropletQC/ | ../output/pilot3.0_iPSC/01_cellcalling-dropletQC |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3.0_MN/01_cellcalling-dropletQC/ | ../output/pilot3.0_MN/01_cellcalling-dropletQC |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version a1fa048. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .cache/
Ignored: .config/
Ignored: .nv/
Ignored: .snakemake/
Ignored: BAUH_2020_MND-single-cell.Rproj
Ignored: GRCh38_turboGFP-RFP_reference/
Ignored: Homo_sapiens.GRCh38.turboGFP/
Ignored: Rplots.pdf
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/pilot3_aggr-experiments.csv
Ignored: data/pilot3_donors.txt
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/2021-04-27_pilot2_nCells-per-donor.pdf
Ignored: output/2021-08-03_pilot2_nCells-per-donor.pdf
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: output/pilot2_testing/
Ignored: output/pilot3.0/
Ignored: output/pilot3.0_MN/
Ignored: output/pilot3.0_captures-separate/
Ignored: output/pilot3.0_iPSC/
Ignored: output/pilot3_Lenti/
Ignored: references/Homo_sapiens.GRCh38.turboGFP.bed
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa.fai
Ignored: references/Homo_sapiens.GRCh38.turboGFP.filtered.gtf
Ignored: references/Homo_sapiens.GRCh38.turboGFP.gtf
Ignored: references/SAindex/
Ignored: references/geno_test.vcf.gz
Ignored: references/pilot3/
Ignored: references/test
Ignored: references/turboGFP.fa
Ignored: references/turboGFP.gtf
Ignored: references/turboRFP.fa
Ignored: workflow/rules/
Untracked files:
Untracked: Capture5-GEX/
Untracked: __Capture5-GEX.mro
Untracked: analysis/2022-03-01_pilot3_dropkick.Rmd
Untracked: analysis/2022-03-01_pilot3_dropletQC.Rmd
Untracked: cellbender.dockerfile
Untracked: hwe1e-05_maf05_vcf_stats.txt
Untracked: hwe1e-05_vcf_stats.txt
Unstaged changes:
Modified: analysis/index.Rmd
Modified: config/config_pilot3.0_MN.yml
Modified: config/config_pilot3.0_iPSC.yml
Modified: workflow/Snakefile
Modified: workflow/rules_cellcalling.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
DropletQC
A new droplet single-cell QC package from Joseph Powell’s lab that is able to detect empty droplets, damaged, and intact cells, and accurately distinguish from one another. This approach is based on a novel quality control metric, the nuclear fraction, which quantifies for each droplet the fraction of RNA originating from unspliced, nuclear pre-mRNA ( preprint).
The nuclear fraction is calculated as:
nuclear fraction = intronic reads / (intronic reads + exonic reads)
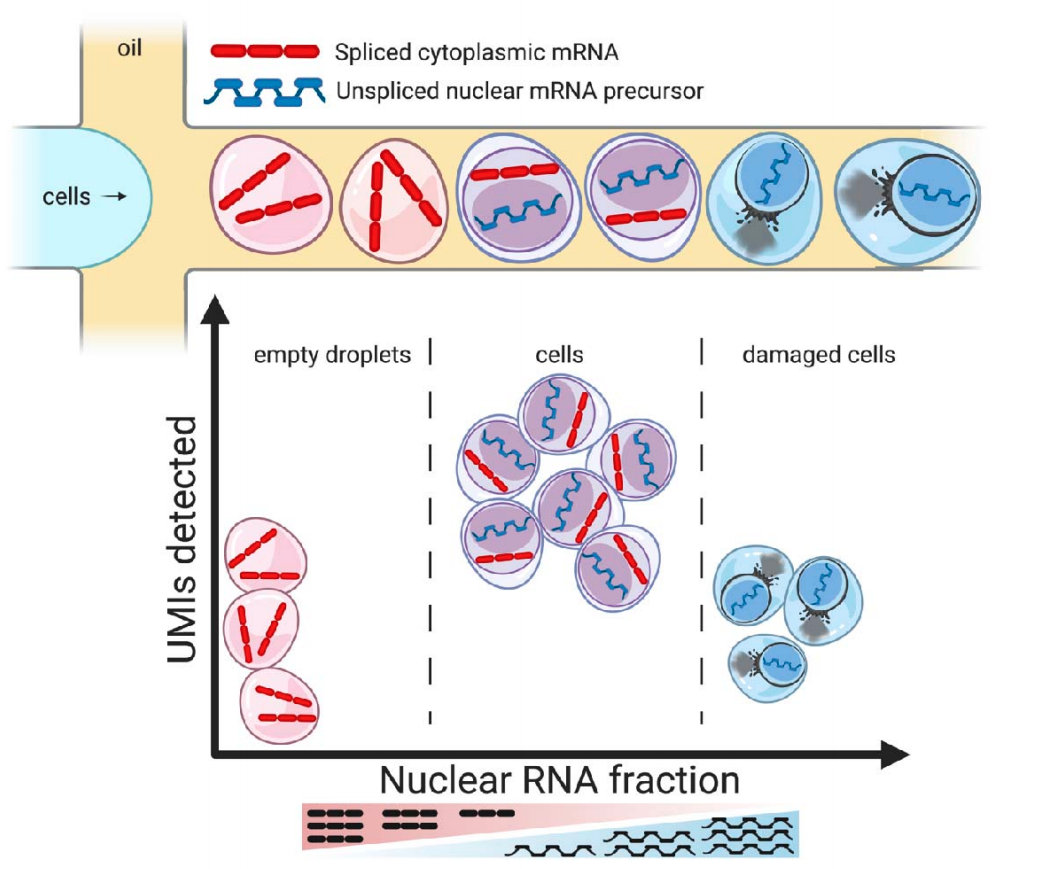
DropletQC theory
Examples from dropletQC paper
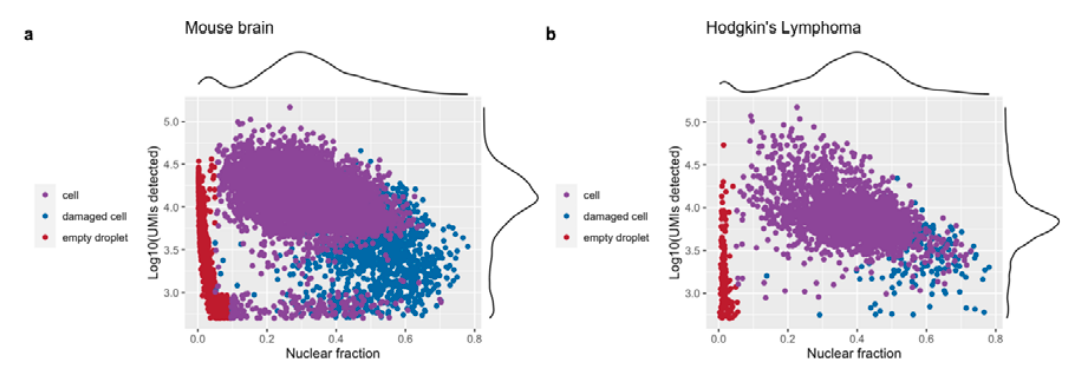
Examples from DropletQC paper
iPSC (Capture5) results
Empty droplets have a low RNA content and low nuclear fraction score (bottom left). Damaged cells have a low RNA content and high nuclear fraction score (bottom right).
c5.stats <- readRDS(paste0(ipsc_dir, "Capture5-GEX/sce_annotated.rds"))
table(c5.stats$df$cell_status)
cell damaged_cell empty_droplet
348940 255078 1001677 ggplot(c5.stats[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_cowplot() +
scale_color_manual(values=list(cell="#784a9f",
damaged_cell="#5280b6",
empty_droplet="#af1f30"))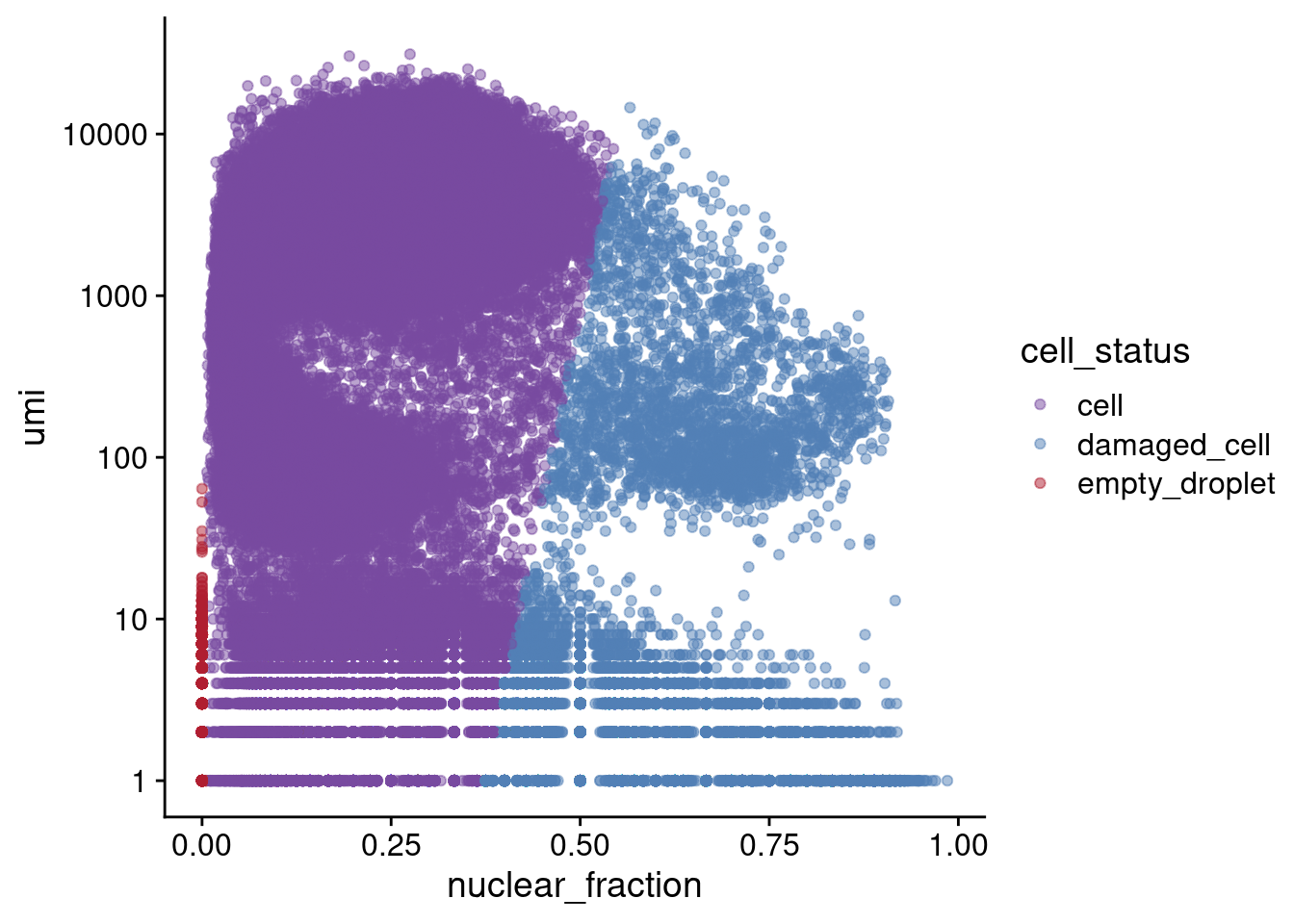
We can also look at the distribution of the nuclear fraction (center) and log10(UMI) counts for cells (BLUE) and damaged cells (RED), where For a population of cells to be called damaged the mean of the distribution fit to the NF for the damaged cells (red line) must be at least nf_sep (default 0.15, threshold at dashed blue line) > the mean of the cell population AND the mean of the distribution fit to the log10(UMI counts) (red line) must be at least umi_sep_perc (default 50%, threshold at dashed blue line) percent < the mean of the cell population.
c5.stats$plots$mn 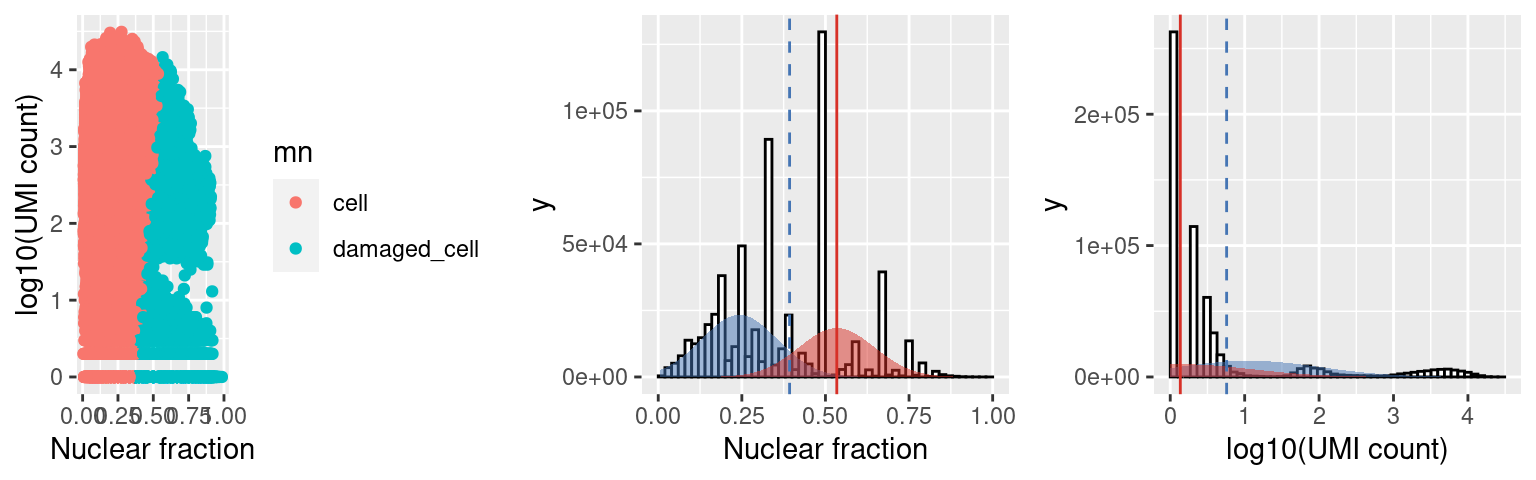
distribution of fit to the nuclear fracation (center) and log10(umi) (right), where cells are shown in blue and damaged are in red (opposite in scalleter plot on the left).
MN (Capture 1-4) results
c1.stats <- readRDS(paste0(mn_dir, "Capture1-GEX/sce_annotated.rds"))
c2.stats <- readRDS(paste0(mn_dir, "Capture2-GEX/sce_annotated.rds"))
c3.stats <- readRDS(paste0(mn_dir, "Capture3-GEX/sce_annotated.rds"))
c4.stats <- readRDS(paste0(mn_dir, "Capture4-GEX/sce_annotated.rds"))
c1.stats$df$capture <- "1"
c2.stats$df$capture <- "2"
c3.stats$df$capture <- "3"
c4.stats$df$capture <- "4"
c5.stats$df$capture <- "5"
stats.dfs <- rbind(c1.stats$df, c2.stats$df, c3.stats$df, c4.stats$df, c5.stats$df)
table(stats.dfs$capture, stats.dfs$cell_status)
cell damaged_cell empty_droplet
1 100116 412886 490872
2 338887 256062 547994
3 501074 0 480747
4 332017 230644 594098
5 348940 255078 1001677Generally, dropletQC results in weird cell calls, where for some captures there are clear minimum umi counts (capture 1) and for others there are no cells determined to be damaged (capture 3). To better use these data to call cells, some guided clustering may be helpful!
p1 <- ggplot(c1.stats[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_cowplot() +
scale_color_manual(values=list(cell="#784a9f",
damaged_cell="#5280b6",
empty_droplet="#af1f30"))
p2 <- ggplot(c2.stats[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_cowplot() +
scale_color_manual(values=list(cell="#784a9f",
damaged_cell="#5280b6",
empty_droplet="#af1f30"))
p3 <- ggplot(c3.stats[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_cowplot() +
scale_color_manual(values=list(cell="#784a9f",
damaged_cell="#5280b6",
empty_droplet="#af1f30"))
p4 <- ggplot(c4.stats[[1]], aes(x=nuclear_fraction, y=umi, color=cell_status)) +
geom_point(alpha=0.5) + scale_y_continuous(trans="log10") + theme_cowplot() +
scale_color_manual(values=list(cell="#784a9f",
damaged_cell="#5280b6",
empty_droplet="#af1f30"))
legend <- get_legend(p1 + theme(legend.box.margin = margin(0, 0, 0, 12)))
plots <- plot_grid(p1 + theme(legend.position="none"), p2 + theme(legend.position="none"),
p3 + theme(legend.position="none"), p4 + theme(legend.position="none"),
nrow=2)
plot_grid(plots, legend, ncol=2, rel_widths = c(7, 1))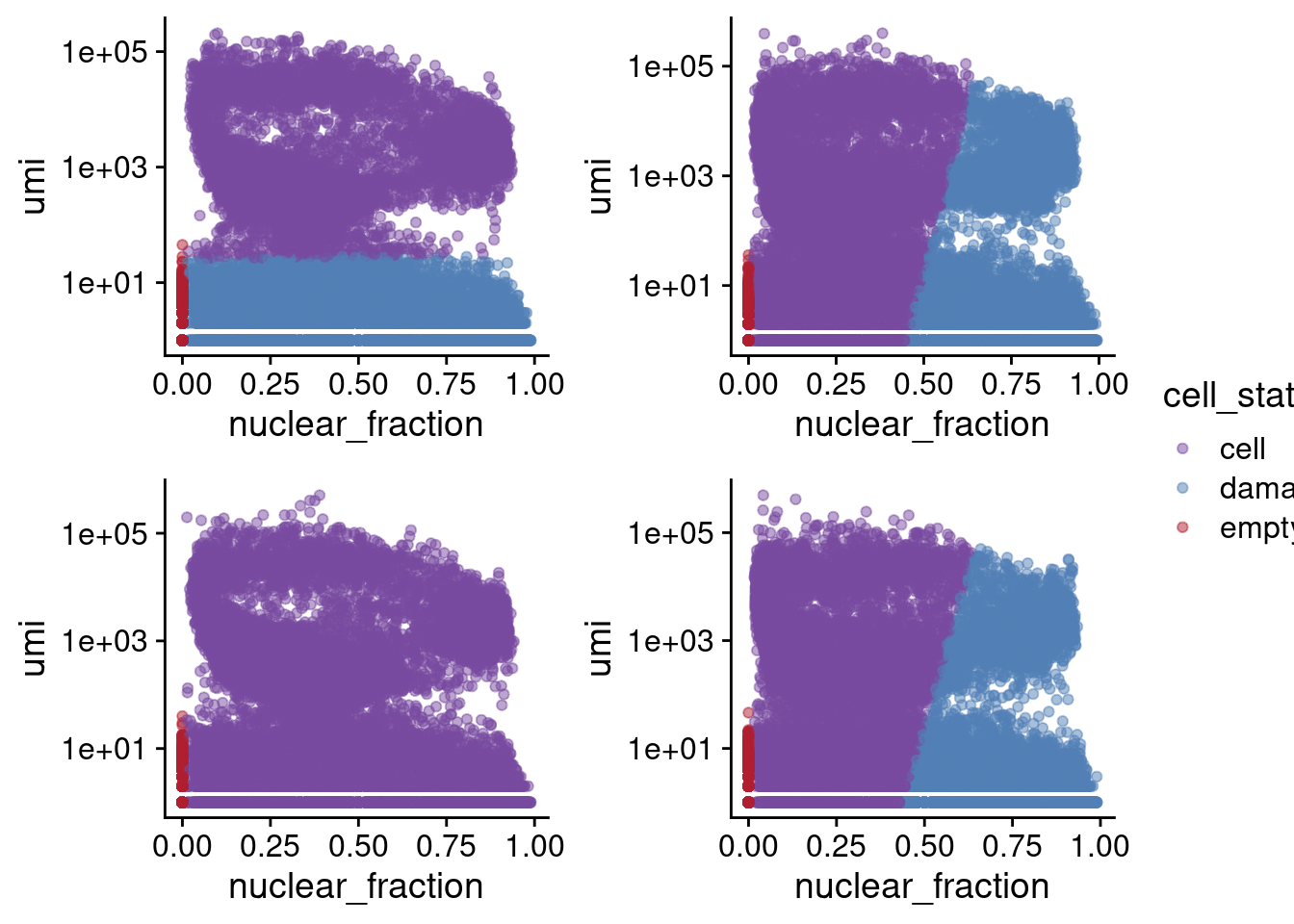
Distribution plots
c1.stats$plots$mn 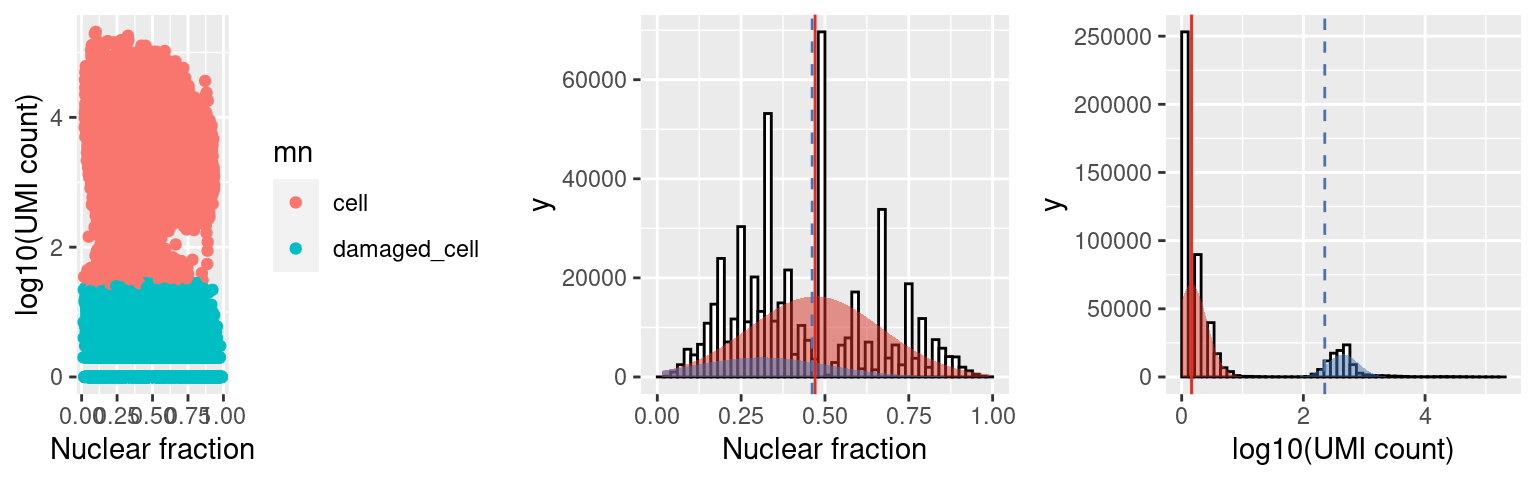
Capture 1
c2.stats$plots$mn 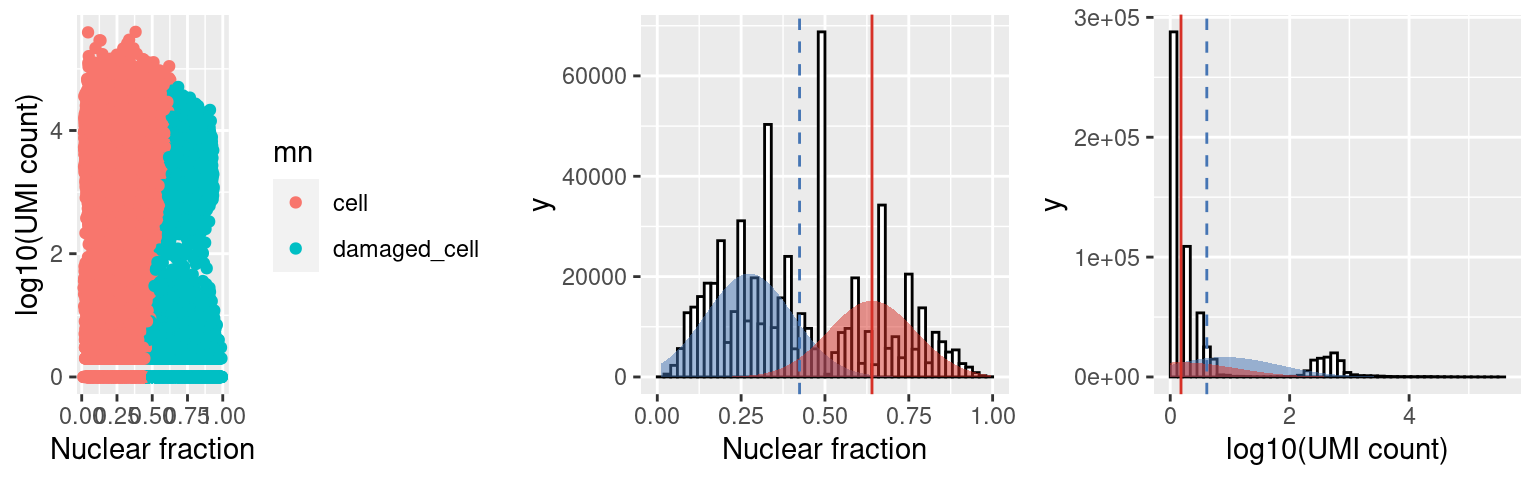
Capture 2
c3.stats$plots$mn 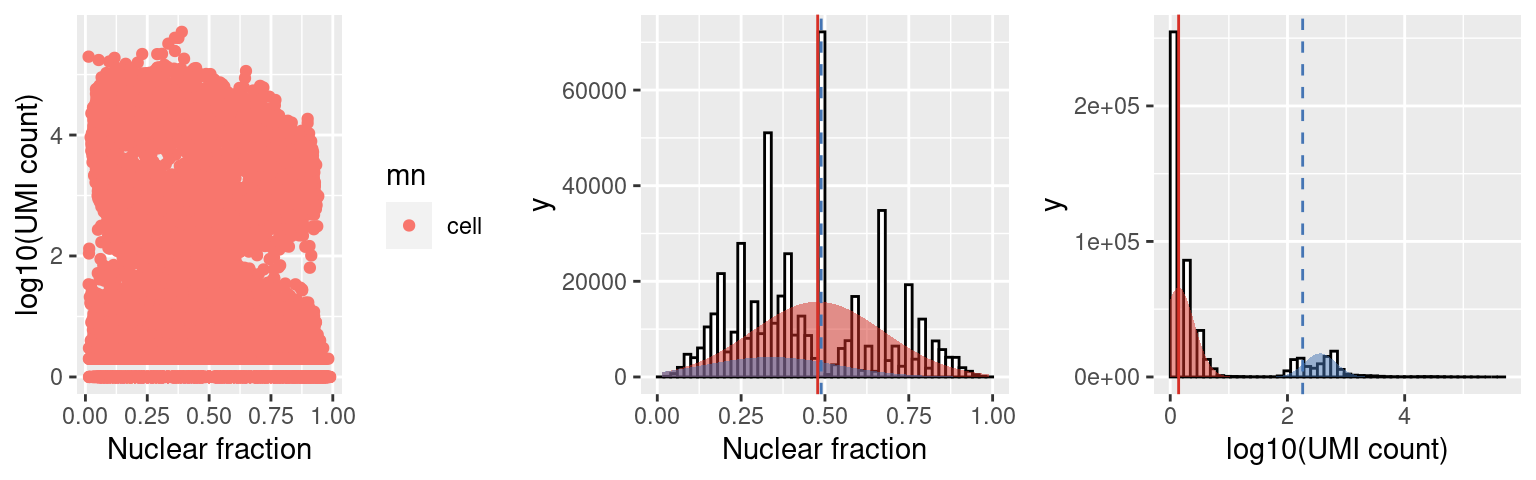
Capture 3
c4.stats$plots$mn 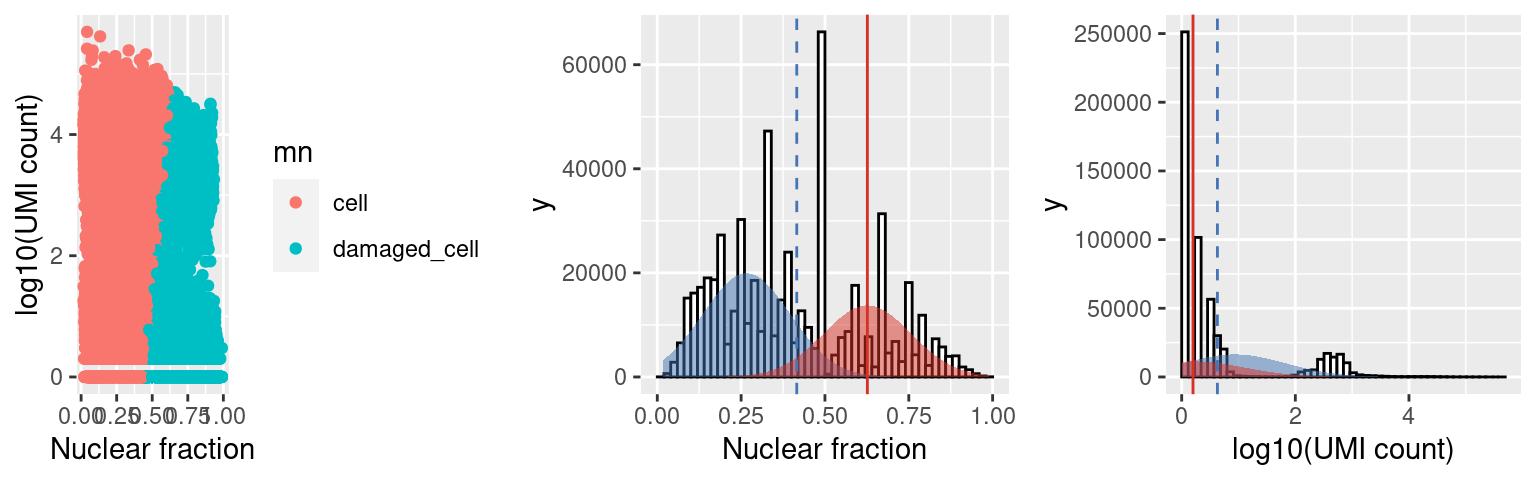
Capture 4
Reasonable umi thresholding
dropletQC does not perform any strict umi filtering and we are getting 10ks of barcodes called as cells with very small umi counts. Short of clustering and choosing clusters by hand, there isn’t a way using dropletQC to hone in on higher confidence cells, so for now I will just look for a minimum UMI count threshold, that when applied gives me about the number of cells we are hoping to recover in each capture (~15k). That threshold is 700 umi:
stats.dfs$cell_status2 <- stats.dfs$cell_status
stats.dfs[stats.dfs$umi < 700 & stats.dfs$cell_status == "cell",
"cell_status2"] <- "empty_droplet"
table(stats.dfs$capture, stats.dfs$cell_status2)
cell damaged_cell empty_droplet
1 11747 412886 579241
2 14070 256062 872811
3 15170 0 966651
4 16573 230644 909542
5 53695 255078 1296922
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Red Hat Enterprise Linux 8.5 (Ootpa)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] ComplexHeatmap_2.10.0 ggvenn_0.1.9 ggplot2_3.3.5
[4] data.table_1.14.2 tidyr_1.1.4 dplyr_1.0.7
[7] cowplot_1.1.1
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 circlize_0.4.13 png_0.1-7
[4] assertthat_0.2.1 rprojroot_2.0.2 digest_0.6.29
[7] foreach_1.5.1 utf8_1.2.2 R6_2.5.1
[10] stats4_4.1.1 evaluate_0.14 highr_0.9
[13] pillar_1.6.4 GlobalOptions_0.1.2 rlang_0.4.12
[16] jquerylib_0.1.4 S4Vectors_0.32.3 GetoptLong_1.0.5
[19] rmarkdown_2.11 labeling_0.4.2 stringr_1.4.0
[22] munsell_0.5.0 compiler_4.1.1 httpuv_1.6.5
[25] xfun_0.28 pkgconfig_2.0.3 BiocGenerics_0.40.0
[28] shape_1.4.6 htmltools_0.5.2 tidyselect_1.1.1
[31] tibble_3.1.6 workflowr_1.6.2 IRanges_2.28.0
[34] codetools_0.2-18 matrixStats_0.61.0 fansi_1.0.0
[37] crayon_1.4.2 withr_2.4.3 later_1.3.0
[40] jsonlite_1.7.2 gtable_0.3.0 lifecycle_1.0.1
[43] DBI_1.1.1 git2r_0.29.0 magrittr_2.0.1
[46] scales_1.1.1 stringi_1.7.6 farver_2.1.0
[49] fs_1.5.2 promises_1.2.0.1 doParallel_1.0.16
[52] bslib_0.3.1 ellipsis_0.3.2 generics_0.1.1
[55] vctrs_0.3.8 rjson_0.2.20 RColorBrewer_1.1-2
[58] iterators_1.0.13 tools_4.1.1 glue_1.6.0
[61] purrr_0.3.4 parallel_4.1.1 fastmap_1.1.0
[64] yaml_2.2.1 clue_0.3-60 colorspace_2.0-2
[67] cluster_2.1.2 knitr_1.36 sass_0.4.0