Lenti barcode analysis
Christina Azodi
2022-10-01
Last updated: 2022-10-01
Checks: 5 2
Knit directory:
BAUH_2020_MND-single-cell/analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(12345) was run prior to running the
code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot4.0_Lenti/01_fasta/ | ../output/pilot4.0_Lenti/01_fasta |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot4.0_Lenti/02_blast/ | ../output/pilot4.0_Lenti/02_blast |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot3_Lenti/02_blast/ | ../output/pilot3_Lenti/02_blast |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot4.0_Lenti/03_keys/ | ../output/pilot4.0_Lenti/03_keys |
| /mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot4.0_iPSC_incIntrons/03_vireo-TX/C099_iPSC_GEX/donor_ids.tsv | ../output/pilot4.0_iPSC_incIntrons/03_vireo-TX/C099_iPSC_GEX/donor_ids.tsv |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version a9c8ba5. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .cache/
Ignored: .config/
Ignored: .snakemake/
Ignored: 20220422_tSNE_plots.pdf
Ignored: BAUH_2020_MND-single-cell.Rproj
Ignored: GRCh38_turboGFP-RFP_reference/
Ignored: Homo_sapiens.GRCh38.turboGFP/
Ignored: Rplots.pdf
Ignored: analysis/2022-04-22_pilot2_CelltypeAbundance.pdf
Ignored: analysis/2022-07-14_pilot4_analysis_part1.pdf
Ignored: code/.ipynb_checkpoints/
Ignored: data/1-s2.0-S0002929720300781-main.pdf
Ignored: data/2103.11251.pdf
Ignored: data/3M-february-2018.txt
Ignored: data/737K-august-2016.txt
Ignored: data/Cellecta-SEQ-CloneTracker-XP_14bp_barcodes.txt
Ignored: data/Cellecta-SEQ-CloneTracker-XP_30bp_barcodes.txt
Ignored: data/STAR_index/
Ignored: data/STAR_output/
Ignored: data/donor_metadata.txt
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.log
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.recode.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.sort.vcf.gz.csi
Ignored: data/genome1K.phase3.SNP_AF5e2.chr1toX.hg38.vcf.gz
Ignored: data/genome1k.chr22.log
Ignored: data/genome1k.chr22.recode.vcf
Ignored: data/pilot2_donors.txt
Ignored: data/pilot3_aggr-experiments.csv
Ignored: data/pilot3_donors.txt
Ignored: data/pilot3_lenti_barcodes_capture5_poolA_D42pass.txt
Ignored: data/s41588-018-0268-8.pdf
Ignored: data/tr2g_hs.tsv
Ignored: logs/
Ignored: output/2021-04-27_pilot2_nCells-per-donor.pdf
Ignored: output/2021-08-03_pilot2_nCells-per-donor.pdf
Ignored: output/CB-scRNAv31-GEX-lib01_QC_metadata.txt
Ignored: output/CB-scRNAv31-GEX-lib02_QC_metadata.txt
Ignored: output/pilot1_starsoloED/
Ignored: output/pilot2.1_gex/
Ignored: output/pilot2_HTO-2/
Ignored: output/pilot2_HTO/
Ignored: output/pilot2_gex_MAF01-152/
Ignored: output/pilot2_gex_MAF01/
Ignored: output/pilot2_gex_starsolo/
Ignored: output/pilot2_gex_starsoloED_GFP/
Ignored: output/pilot3.0_MN/
Ignored: output/pilot3.0_iPSC/
Ignored: output/pilot3.1_iPSC/
Ignored: output/pilot3_Lenti/
Ignored: output/pilot4.0_Lenti/
Ignored: output/pilot4.0_iPSC/
Ignored: output/pilot4.0_iPSC_incIntrons/
Ignored: output/pilot4.0_nuclei/
Ignored: output/pilot4.0_nuclei_incIntrons/
Ignored: references/Homo_sapiens.GRCh38.turboGFP.bed
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa
Ignored: references/Homo_sapiens.GRCh38.turboGFP.fa.fai
Ignored: references/Homo_sapiens.GRCh38.turboGFP.filtered.gtf
Ignored: references/Homo_sapiens.GRCh38.turboGFP.gtf
Ignored: references/Homo_sapiens.GRCh38.turboGFP_gene.gtf
Ignored: references/SAindex/
Ignored: references/allen_brain_reference/
Ignored: references/geno_test.vcf.gz
Ignored: references/pilot3/
Ignored: references/pilot3_20220330/
Ignored: references/pilot4/
Ignored: references/test
Ignored: references/turboGFP.fa
Ignored: references/turboGFP.gtf
Ignored: references/turboRFP.fa
Ignored: workflow/rules/
Untracked files:
Untracked: .nv/
Untracked: analysis/2022-07-14_pilot4_analysis.Rmd
Untracked: analysis/2022-07-14_pilot4_analysis_part1.Rmd
Untracked: analysis/2022-08-22_pilot4_analysis_part2.Rmd
Untracked: analysis/2022-09-12_pilot4_LentiBarcodes.Rmd
Untracked: analysis/2022-09-12_pilot4_LentiBarcodes.log
Untracked: analysis/2022-09-12_pilot4_LentiBarcodes.tex
Untracked: cellbender.dockerfile
Untracked: config/config_pilot4.0_Lenti.yml
Untracked: config/config_pilot4.0_iPSC.yml
Untracked: config/config_pilot4.0_nuclei.yml
Untracked: hwe1e-05_maf05_vcf_stats.txt
Untracked: hwe1e-05_vcf_stats.txt
Untracked: workflow/Snakefile_Lenti_pilot4.smk
Untracked: workflow/Snakefile_simple.smk
Untracked: workflow/vireo-p4_rm197 copy.sh
Untracked: workflow/vireo-p4_rm197.sh
Unstaged changes:
Modified: analysis/2022-05-03_pilot3_Cell-demultiplexing-Lenti.Rmd
Modified: analysis/index.Rmd
Modified: config/config_pilot3.0_MN.yml
Modified: config/config_pilot3.0_iPSC.yml
Modified: workflow/demuxlet_jobs/demux_capture5.sh
Modified: workflow/rules_demultiplexing.smk
Modified: workflow/rules_prepare-genotype-data.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Goal
Attribute lenti barcodes (experiment S000312; R1=cell barcodes; R2=lenti barcodes) with iPSC cell barcodes/demultiplexed donor IDs (pilot #4 exp ID: C099).
Blastn results

blastn strategy
Example scripts:
14 bp barcode: blastn -db output/pilot4_Lenti/00_sequences/bc14_forward.fa -query output/pilot4_Lenti/01_fasta/S000312-Lenti.fa -query_loc 26-40 -word_size 5 -max_hsps 1 -evalue 0.1 -num_threads 4 -outfmt 6 -out output/pilot4_Lenti/02_blast/S000312-Lenti_bc14_blast.out
30 bp barcode: blastn -db output/pilot4_Lenti/00_sequences/bc30_forward.fa -query output/pilot4_Lenti/01_fasta/S000312-Lenti.fa -query_loc 46-76 -word_size 10 -max_hsps 1 -evalue 0.1 -max_target_seqs 1 -outfmt 6 -out output/pilot4_Lenti/02_blast/S000312-Lenti_bc30_blast.out
Read in blastn hits and remove ambiguous matches by first keeping the longest hit for each read and if two lenti barcodes have equally long hits, removing the read entirely. Number of reads with lenti barcode matches:
load_parse_blastn <- function(path) {
df <- fread(path, header=FALSE, select = c(1:6))
names(df) <- c("qseqid", "sseqid", "pident", "length", "mismatch", "gapopen")
# If >1 hit for a lenti barcode, keep the one(s) with the longer overlap
df <- df[df[, .I[length == max(length)], by=qseqid]$V1]
# If >1 hit for a lenti barcode with equal lengths, remove them both.
df <- df[!(duplicated(df$qseqid) | duplicated(df$qseqid, fromLast = TRUE)), ]
return(df)
}
## Load and process blastn results
bc_header <- load_parse_blastn(paste0(blast_out, "S000312_header_blast.out"))
p3_c4_header <- load_parse_blastn(paste0(blast_out_p3, "Capture4-Lenti_header_blast.out"))
p3_c5_header <- load_parse_blastn(paste0(blast_out_p3, "Capture5-Lenti_header_blast.out"))
p1 <- gghistogram(bc_header, x="length", bins=27)
p2 <- gghistogram(p3_c4_header, x="length", bins=27)
p3 <- gghistogram(p3_c5_header, x="length", bins=27)
plot_grid(p1, p2, p3, ncol=3, labels="auto")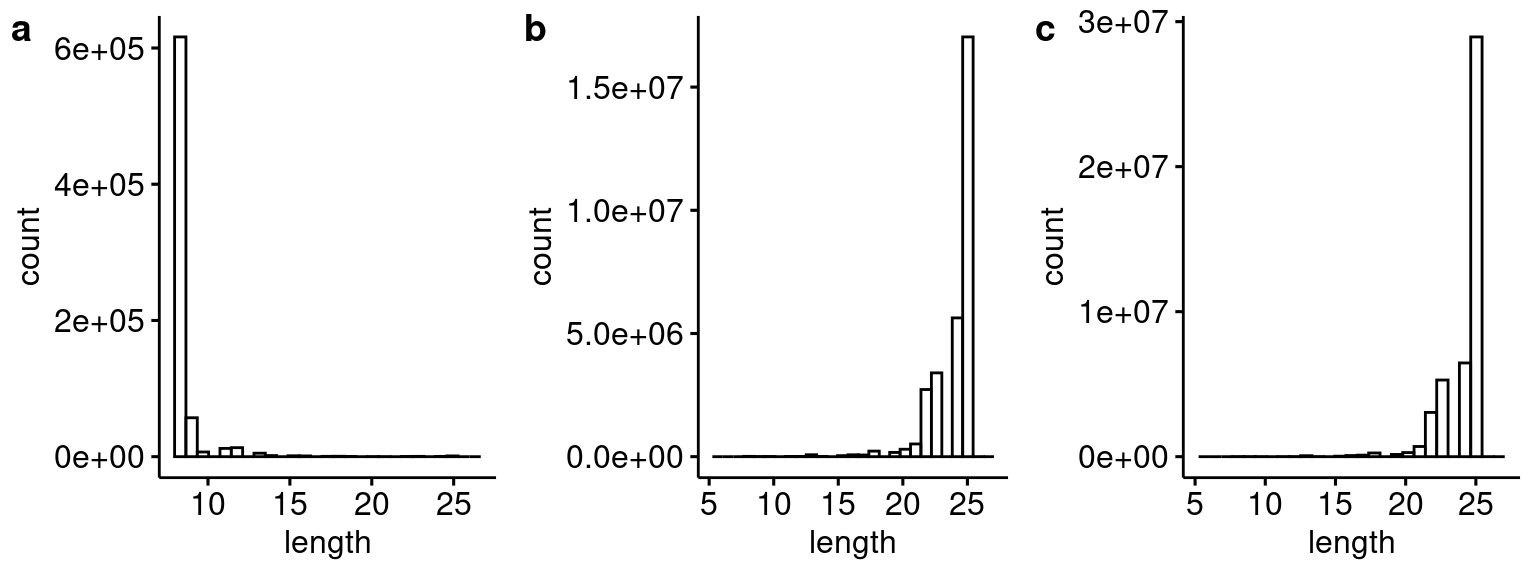
Distribution of blastn hit lengths for the 25 bp header sequence for (a) this analysis compared to (b) Capture 4 and (c) Capture 5 from the first Lenti-barcode test (i.e. pilot 3).
rm(p3_c4_header, p3_c5_header)Compared to the last Lenti barcode analysis, these reads have very low quality headers.
bc_bc14 <- load_parse_blastn(paste0(blast_out, "S000312_bc14_blast.out"))
p3_bc14 <- load_parse_blastn(paste0(blast_out_p3, "Capture5-Lenti_bc14_blast.out"))
p1 <- gghistogram(bc_header, x="length", bins=15)
p2 <- gghistogram(p3_bc14, x="length", bins=15)
plot_grid(p1, p2, ncol=2, labels="auto")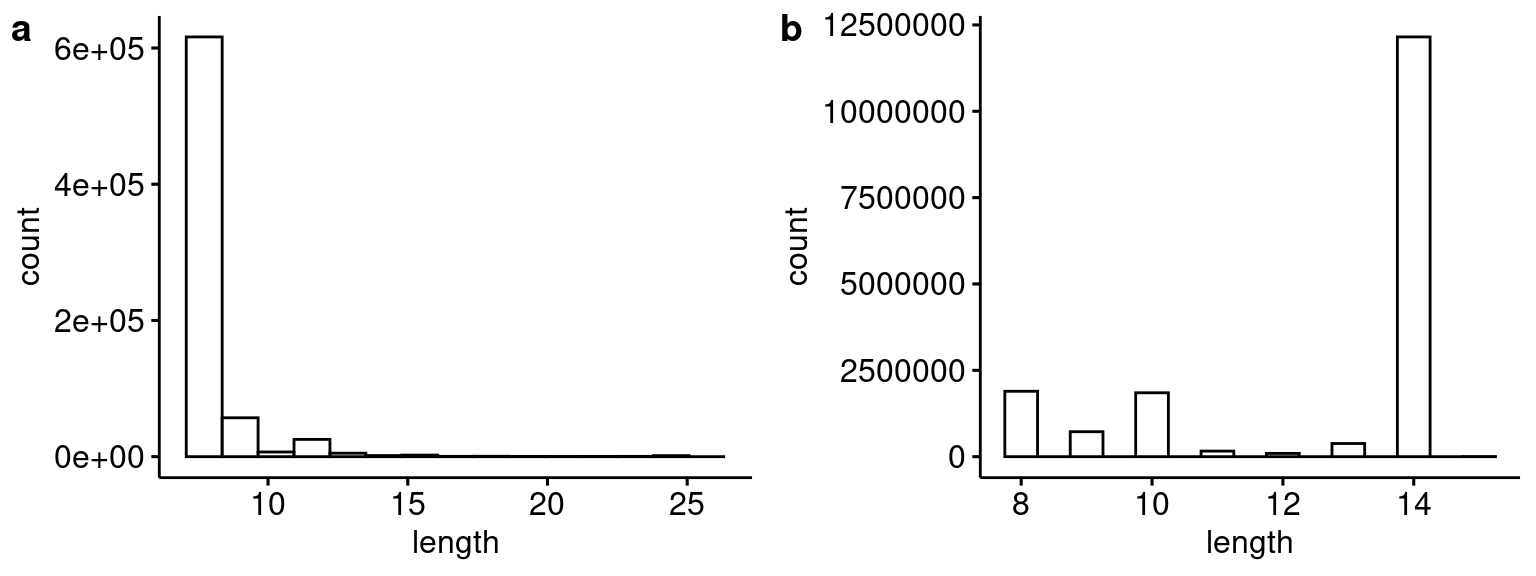
Distribution of blastn hit lengths for the 14 bp barcode sequence for (a) this analysis compared to (b) Capture 5 from the first Lenti-barcode test (i.e. pilot 3).
rm(p3_bc14)bc_bc30 <- load_parse_blastn(paste0(blast_out, "S000312_bc30_blast.out"))
p3_bc30<- load_parse_blastn(paste0(blast_out_p3, "Capture5-Lenti_bc30_blast.out"))
p1 <- gghistogram(bc_bc30, x="length", bins=32)
p2 <- gghistogram(p3_bc30, x="length", bins=32)
plot_grid(p1, p2, ncol=2, labels="auto")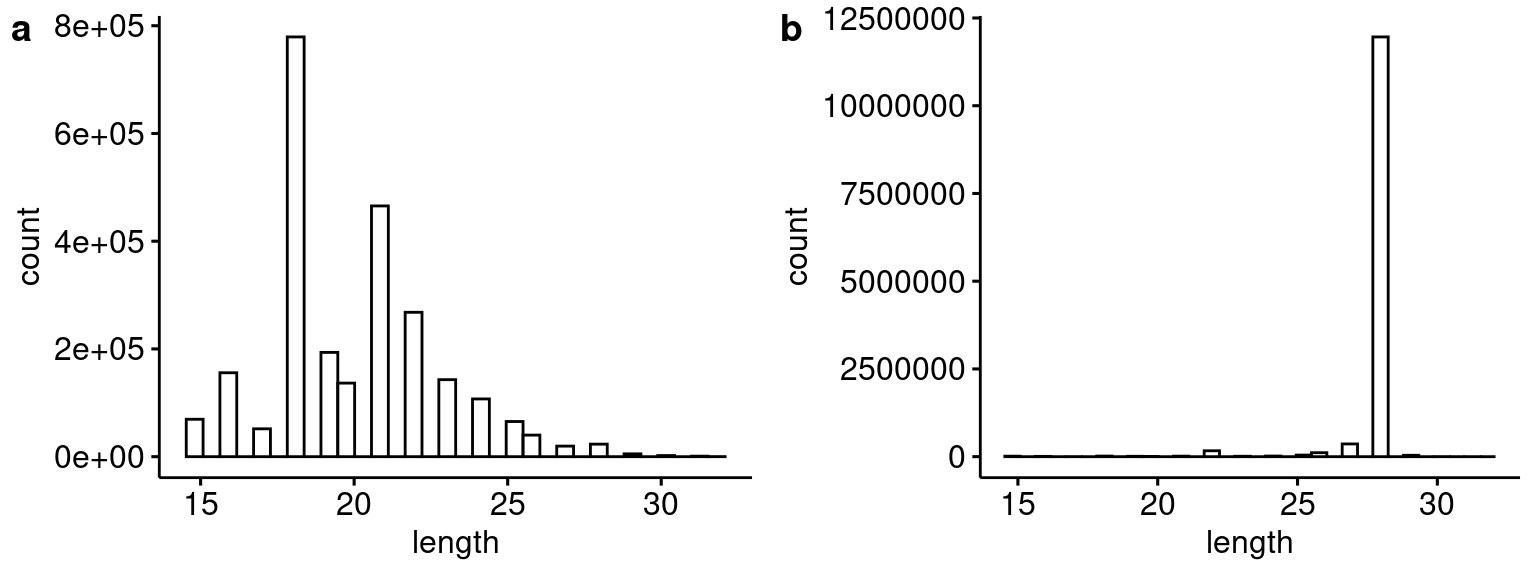
Distribution of blastn hit lengths for the 30 bp barcode sequence for (a) this analysis compared to (b) Capture 5 from the first Lenti-barcode test (i.e. pilot 3).
rm(p3_bc30)Again, in the first test the 14 and 30 bp barcode blast hits were consistently longer than the hits for the current analysis.
# Merge
n_reads <- 474065418
S000312_both <- bc_bc14 %>% inner_join(bc_bc30, by = "qseqid", suffix = c(".bc14", ".bc30"))
message("Of the ", n_reads,
" sequenced, the number with lenti barcode matches (no additional filtering) for: \nbc14: ",
nrow(bc_bc14), " (", round(nrow(bc_bc14)/n_reads*100, 4), "%)",
"\nbc30: ", nrow(bc_bc30), " (", round(nrow(bc_bc30)/n_reads*100, 4), "%)",
"\nBoth bc14 and bc30: ", nrow(S000312_both), " (",
round(nrow(S000312_both)/n_reads*100, 4), "%)")Of the 474065418 sequenced, the number with lenti barcode matches (no additional filtering) for:
bc14: 69894072 (14.7436%)
bc30: 2526639 (0.533%)
Both bc14 and bc30: 435375 (0.0918%)rm(bc_bc14, bc_bc30, bc_header)
gc() used (Mb) gc trigger (Mb) max used (Mb)
Ncells 49237319 2629.6 205192234 10958.5 256490292 13698.1
Vcells 878264420 6700.7 2939757056 22428.6 3659485460 27919.7Note that in the pilot 3 analysis for capture 5 there were 46,041,484 reads, 17,260,005 with bc14 hits, 12,795,040 with bc30 hits… with 12,544,555 reads having a hit for both bc14 and bc30 (27% of total reads).
h1 <- gghistogram(S000312_both, x = "length.bc14")Warning: Using `bins = 30` by default. Pick better value with the argument
`bins`.h2 <- gghistogram(S000312_both, x = "length.bc30")Warning: Using `bins = 30` by default. Pick better value with the argument
`bins`.h_plots <- plot_grid(h1, h2, nrow=1)
p3 <- ggboxplot(S000312_both, x="length.bc14", y = "length.bc30", outlier.shape = NA)
plot_grid(h_plots, p3, ncol=1, labels=c("", "c"))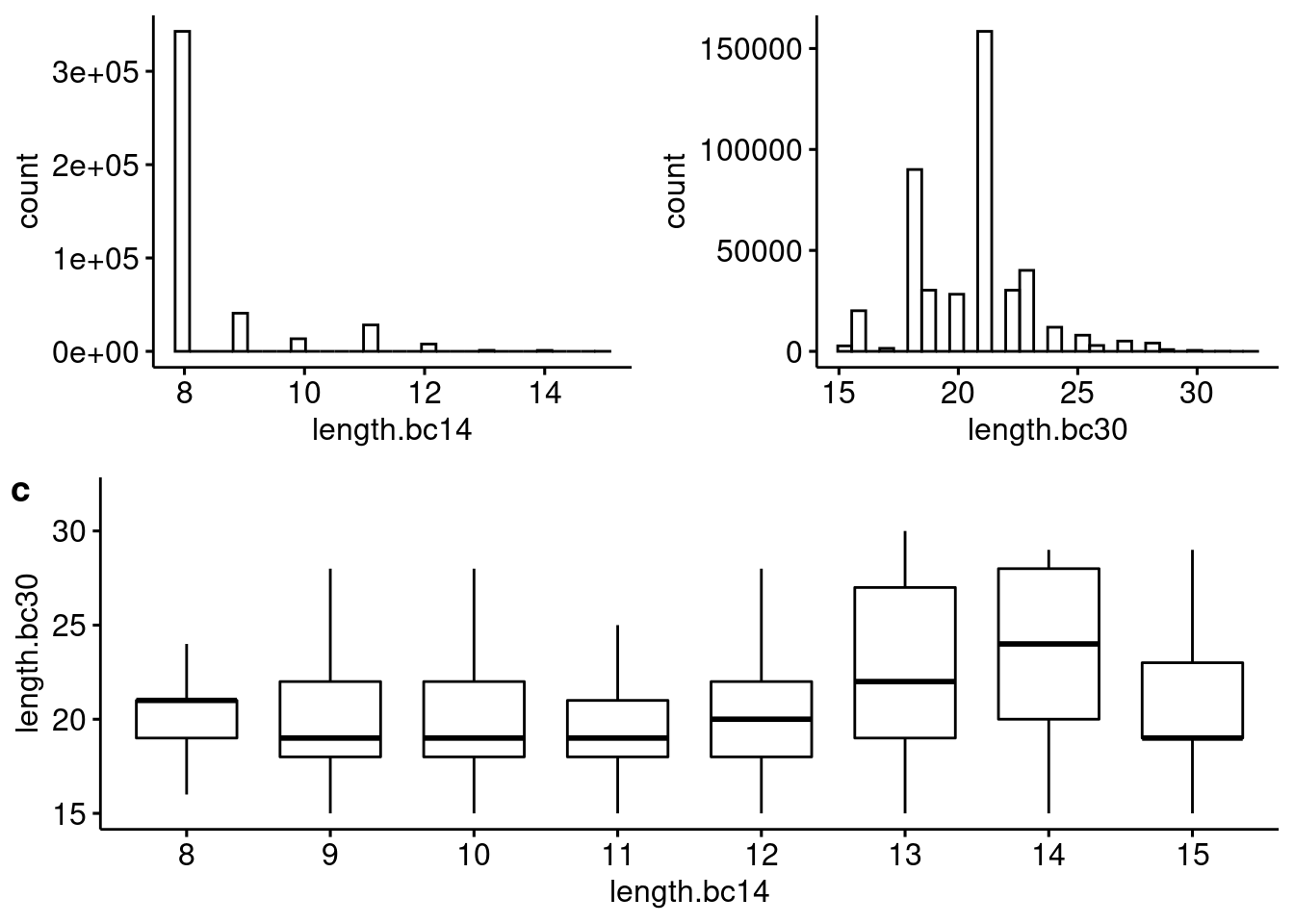
Distribution of (a) bc14 and (b) bc30 blast hit lengths and (c) the relationship between the two across reads.
Filter and merge with cell barcodes
For the last pilot I used “strict” minimum hit length cutoffs of 12 and 26 and an max missmatch rate of 1 and 2 for the bc14 and bc30 blast hits, respectively. However given the lower quality I’m going to be more lenient for this analysis and require lengths 10 and 22 and no max mismatch rate.
# Add lenti barcode IDs
S000312_both$lenti_bc <- paste0("lenti_", S000312_both$sseqid.bc14, "-", S000312_both$sseqid.bc30)
# Filtering by minimum length
S000312_both_filt <- subset(S000312_both, S000312_both$length.bc14 >= 10 & S000312_both$length.bc30 >= 22)
message("Number of barcodes meeting this relatively loose threshold: ", nrow(S000312_both_filt))Number of barcodes meeting this relatively loose threshold: 14365Note that in the first analysis, 12,411,301 reads from capture 5 met this criteria.
Merge with cell barcodes from the R1 reads
The R1 reads are all exactly 28 bp and contain the 16 bp cell barcode followed by a 12 bp UMI.
parse_merge_R1_cellbarcodes <- function(path, lenti) {
cbc <- fread(path, header=FALSE, sep="\t")
summary(nchar(cbc$cell_barcode))
names(cbc) <- c("qseqid", "R1")
cbc$qseqid <- gsub(">", "", gsub(" .*", "", cbc$qseqid))
cbc <- separate(cbc, R1, into = c("cell_barcode", "umi"), sep = 16)
lenti <- lenti %>% left_join(cbc, by = "qseqid")
rm(cbc)
return(lenti)
}
if(rerun){
S000312_both_filt <- parse_merge_R1_cellbarcodes("/mnt/mcfiles/Datasets/BAUH_2020_MND-single-cell/pilot4_20220710/LentiBarcodes/S000312/G000099_iPSC_GEX_S1_L001_R1_001.tsv", S000312_both_filt)
write.table(S000312_both_filt, paste0(out, "S000312-Lenti_merged_barcodes.tsv"),
sep = "\t", quote=FALSE, row.names = FALSE)
} else{
S000312_both_filt <- read.table(paste0(out, "S000312-Lenti_merged_barcodes.tsv"),
sep = "\t", header=TRUE)
}Make lenti2cell barcode keys
How many UMIs support each cell barcode to lenti barcode pair….
S000312_both_filt_counts <- S000312_both_filt %>% distinct(cell_barcode, lenti_bc, umi) %>%
group_by(cell_barcode, lenti_bc) %>%
tally()
table(S000312_both_filt_counts$n)
1 2 3 4
10046 118 1 2 Most pairs have just one UMI supporting the match. Because each cell can have more than 1 lenti barcode, we expect multiple lenti barcodes per cell… Here is the distribution of the number of lenti-barcodes matching to each cell barcode:
lenti_barcode_key <- as.list(unstack(rev(S000312_both_filt[, c("cell_barcode", "lenti_bc")])))
hist(unlist(lapply(lenti_barcode_key, FUN=function(x) length(x))),
main = "lenti barcodes per cell", xlab = "# lenti barcodes")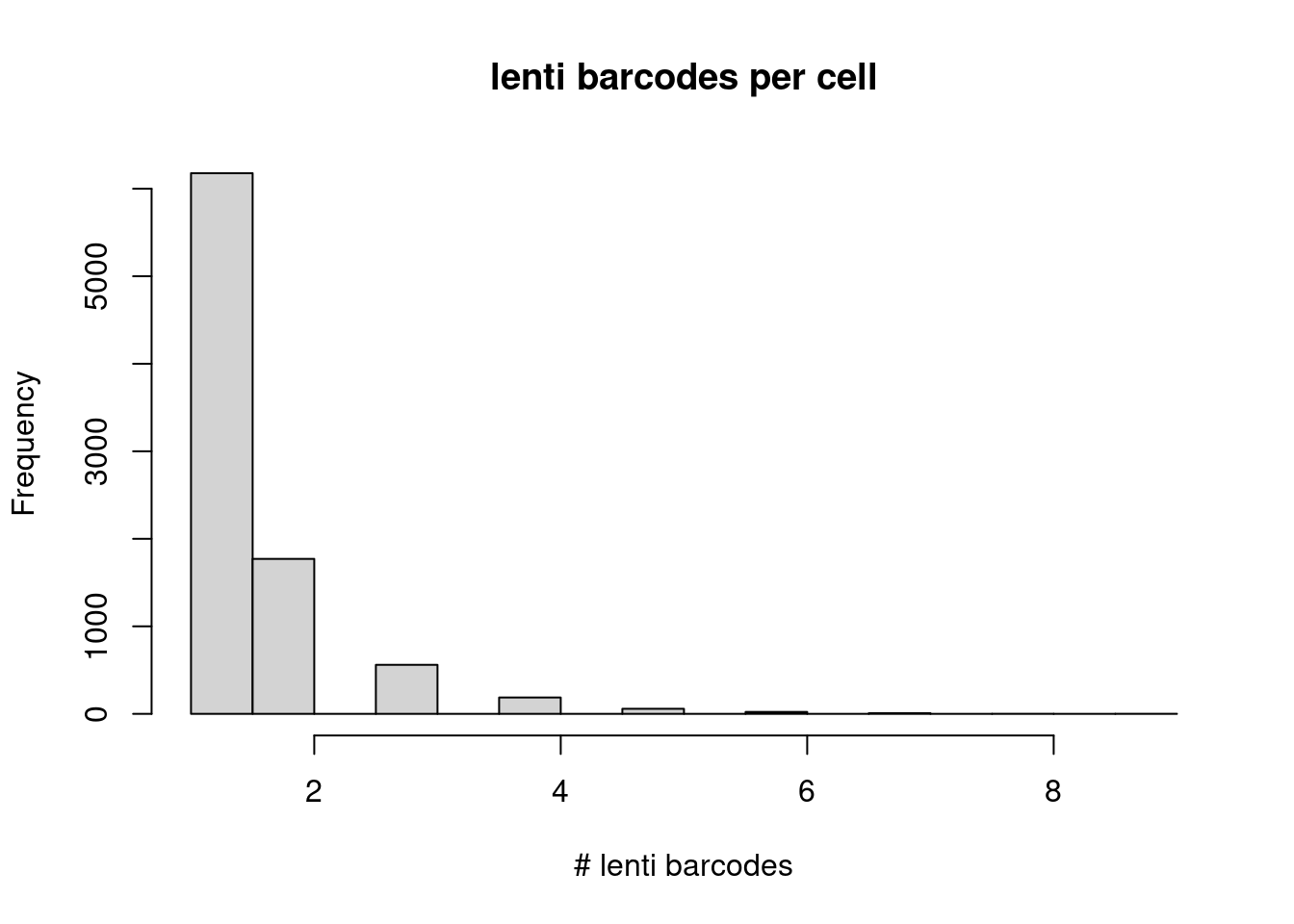
And here is the distribution of the number of cell barcodes matching to each lenti barcode:
lenti_barcode_key_rev <- as.list(unstack(rev(S000312_both_filt[, c("lenti_bc", "cell_barcode")])))
lenti_barcode_key_rev_counts <- unlist(lapply(lenti_barcode_key_rev, FUN=function(x) length(unique(x))))
hist(lenti_barcode_key_rev_counts,
main = "cell barcodes per lenti barcode", xlab = "# cell barcodes")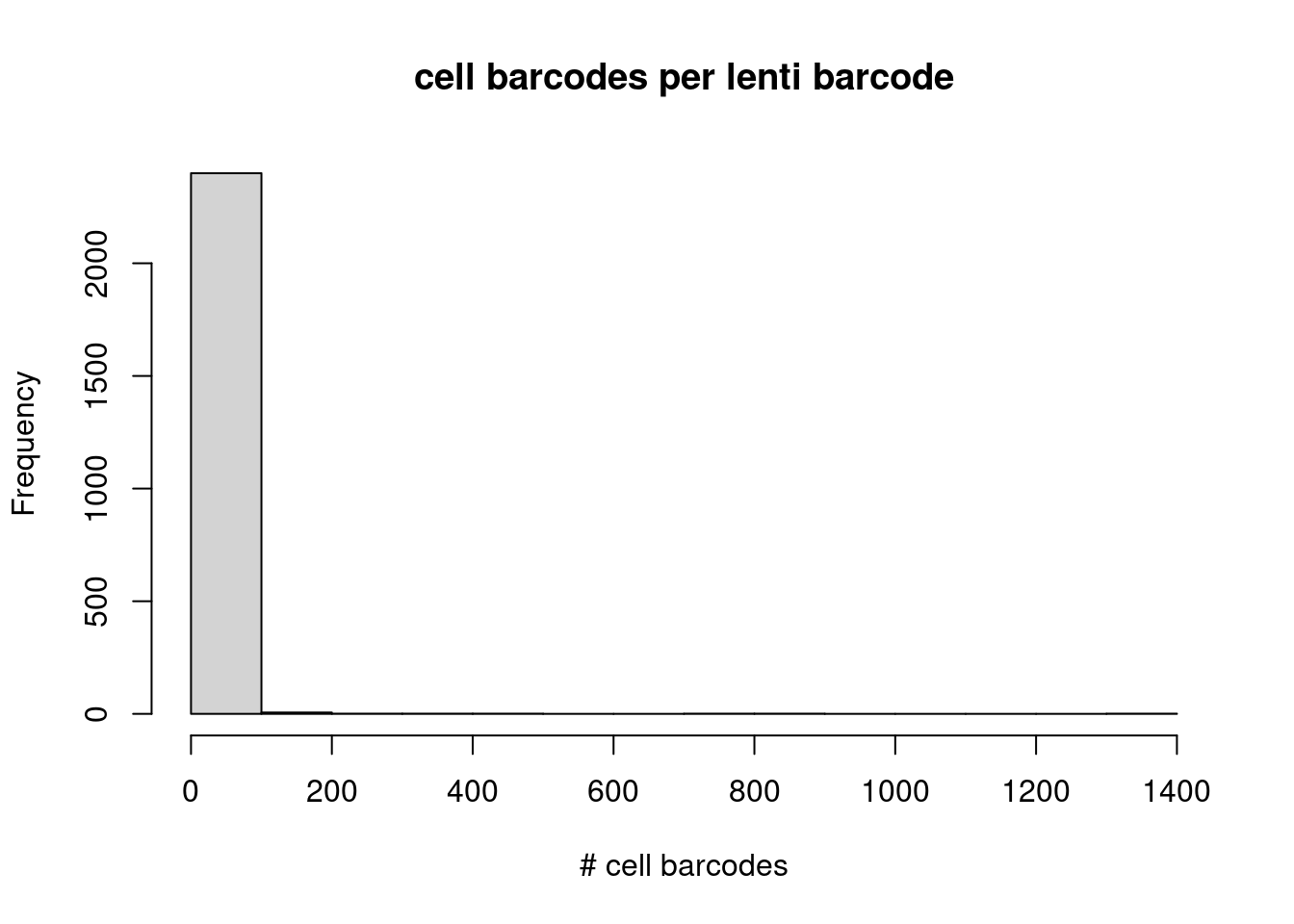
message("Zooming in on lenti-barcodes with > 10 cell barcode matches.")Zooming in on lenti-barcodes with > 10 cell barcode matches.hist(lenti_barcode_key_rev_counts[lenti_barcode_key_rev_counts >10 ],
main = "cell barcodes per lenti barcode", xlab = "# cell barcodes")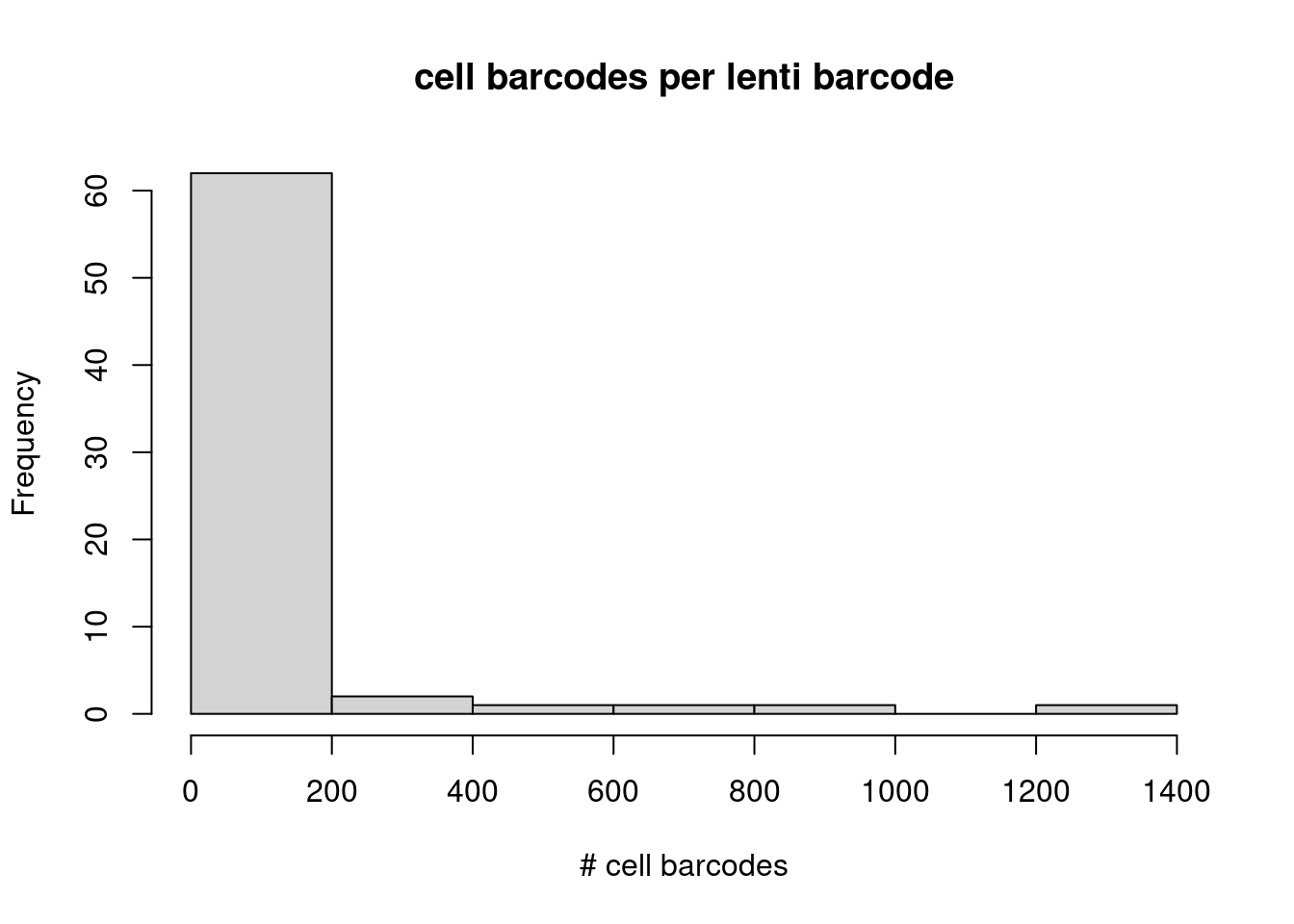
For these Lenti barcodes that match to more than 1 cell barcode, we expect them to be assigned the same donor…
Compare with iPSC donor IDs
p4_ipsc_donors <- read.table("/mnt/beegfs/mccarthy/backed_up/general/cazodi/Projects/BAUH_2020_MND-single-cell/output/pilot4.0_iPSC_incIntrons/03_vireo-TX/C099_iPSC_GEX/donor_ids.tsv", sep="\t", header=TRUE)
p4_ipsc_donors <- subset(p4_ipsc_donors, ! donor_id %in% c("unassigned", "doublet"))
p4_ipsc_donors$cell_barcode <- gsub("-1", "", p4_ipsc_donors$cell)
counts <- table(p4_ipsc_donors$cell_barcode %in% S000312_both_filt$cell_barcode)
message("Of the ", nrow(p4_ipsc_donors), " QCed and donor assigned cells, ",
counts[["TRUE"]],
" (", round(counts[["TRUE"]]/nrow(p4_ipsc_donors)*100, 1),
"%) have lenti-barcode matches.")Of the 10358 QCed and donor assigned cells, 3400 (32.8%) have lenti-barcode matches.How specific are lenti-barcodes to cells assigned to the same donor?
results <- S000312_both_filt %>% left_join(p4_ipsc_donors, by="cell_barcode")
matches <- results[complete.cases(results), ]
matches_key <- as.list(unstack(rev(matches[, c("lenti_bc", "donor_id")])))
matches_key_counts <- unlist(lapply(matches_key, FUN=function(x) length(unique(x))))
hist(matches_key_counts,
main = "donor IDs per lenti barcode", xlab = "# donors")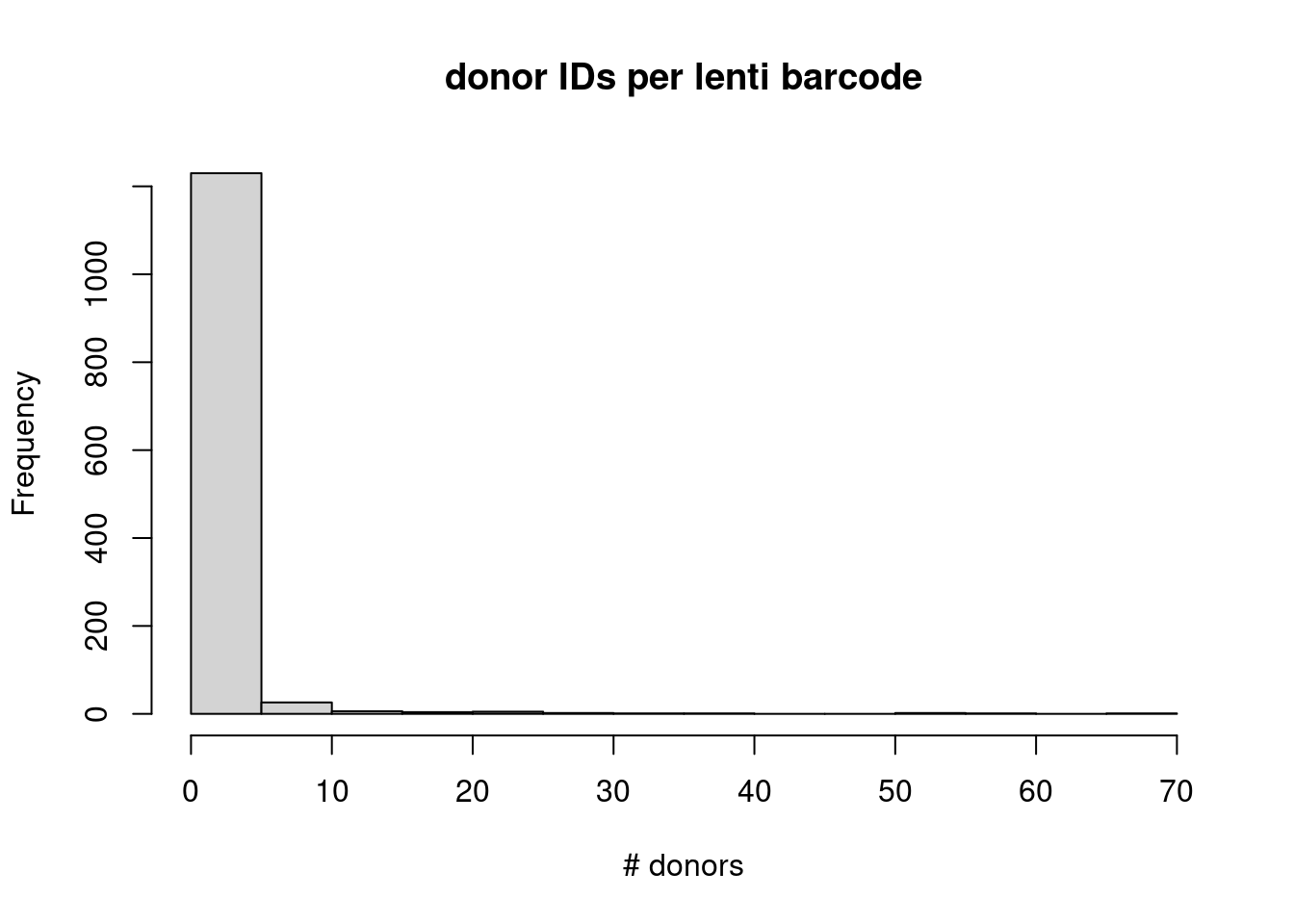
table(matches_key_counts)matches_key_counts
1 2 3 4 5 6 7 8 9 10 11 12 15 16 17 19 21 22 23 25
981 165 50 21 13 6 9 6 4 1 4 1 1 1 1 2 1 2 1 1
27 28 32 38 52 53 57 68
1 1 1 1 1 1 1 1 if(save){write.table(matches, paste0(out, "Lenti2cell_barcode_key.tsv"), sep = "\t",
quote=FALSE, row.names = FALSE)}
sessionInfo()R version 4.1.1 (2021-08-10)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Rocky Linux 8.6 (Green Obsidian)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblasp-r0.3.15.so
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] stringr_1.4.0 viridis_0.6.2 viridisLite_0.4.0 cowplot_1.1.1
[5] ggpubr_0.4.0 ggplot2_3.3.6 data.table_1.14.2 tidyr_1.2.0
[9] dplyr_1.0.9 argparse_2.1.3
loaded via a namespace (and not attached):
[1] tidyselect_1.1.2 xfun_0.30 bslib_0.3.1 purrr_0.3.4
[5] carData_3.0-5 colorspace_2.0-3 vctrs_0.4.1 generics_0.1.3
[9] htmltools_0.5.2 yaml_2.3.5 utf8_1.2.2 rlang_1.0.4
[13] jquerylib_0.1.4 later_1.3.0 pillar_1.7.0 glue_1.6.2
[17] withr_2.5.0 DBI_1.1.3 lifecycle_1.0.1 munsell_0.5.0
[21] ggsignif_0.6.3 gtable_0.3.0 workflowr_1.6.2 evaluate_0.15
[25] labeling_0.4.2 knitr_1.37 fastmap_1.1.0 httpuv_1.6.5
[29] fansi_1.0.3 highr_0.9 broom_0.7.10 Rcpp_1.0.9
[33] backports_1.4.1 promises_1.2.0.1 scales_1.2.0 jsonlite_1.8.0
[37] abind_1.4-5 farver_2.1.1 fs_1.5.2 gridExtra_2.3
[41] digest_0.6.29 stringi_1.7.8 rstatix_0.7.0 rprojroot_2.0.2
[45] grid_4.1.1 cli_3.3.0 tools_4.1.1 magrittr_2.0.3
[49] sass_0.4.0 tibble_3.1.7 car_3.0-12 crayon_1.5.1
[53] pkgconfig_2.0.3 ellipsis_0.3.2 assertthat_0.2.1 rmarkdown_2.11
[57] rstudioapi_0.13 R6_2.5.1 git2r_0.29.0 compiler_4.1.1