10x Fibroblasts: Estimate splatPop parameters
Christina B. Azodi
2021-06-02
Last updated: 2021-06-02
Checks: 6 1
Knit directory: KEJP_2020_splatPop/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210215) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version bd0c8b0. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/.DS_Store
Ignored: data/10x_lung/
Ignored: data/ALL.chr2.phase3_shapeit2_mvncall_integrated_v5a.20130502.genotypes.EURO.0.99.MAF05.filtered.vcf
Ignored: data/D30.h5
Ignored: data/Diabetes/
Ignored: data/IBD/
Ignored: data/agg_10X-fibroblasts-control.rds
Ignored: data/agg_IBD-ss2.rds
Ignored: data/agg_Neuro-10x_DA.rds
Ignored: data/agg_NeuroSeq-10x_D11-pool6-filt.rds
Ignored: data/agg_NeuroSeq-10x_D11-pool6.rds
Ignored: data/agg_diabetes-ss2.rds
Ignored: data/agg_iPSC-ss2_D0.rds
Ignored: data/covid/
Ignored: data/cuomo_NeuroSeq_10x_all_sce.rds
Ignored: data/lung_fibrosis/
Ignored: data/pseudoB_IBD-ss2.rds
Ignored: data/pseudoB_Neuro-10x_DA.rds
Ignored: data/pseudoB_NeuroSeq-10x_D11-pool6-filt.rds
Ignored: data/pseudoB_NeuroSeq-10x_D11-pool6.rds
Ignored: data/pseudoB_diabetes-ss2.rds
Ignored: data/pseudoB_iPSC-ss2_D0.rds
Ignored: data/sce_10X-fibroblasts-947170.rds
Ignored: data/sce_10X-fibroblasts-allGenes.rds
Ignored: data/sce_10X-fibroblasts.rds
Ignored: data/sce_IBD-ss2.rds
Ignored: data/sce_IBD-ss2_HC2.rds
Ignored: data/sce_Neuro-10x.rds
Ignored: data/sce_Neuro-10x_2CT.rds
Ignored: data/sce_Neuro-10x_DA-wihj4.rds
Ignored: data/sce_NeuroSeq-10x_D11-pool6-filt-mita1FPP.rds
Ignored: data/sce_NeuroSeq-10x_D11-pool6-filt.rds
Ignored: data/sce_NeuroSeq-10x_D11-pool6-mita1FPP.rds
Ignored: data/sce_NeuroSeq-10x_D11-pool6.rds
Ignored: data/sce_diabetes-ss2.rds
Ignored: data/sce_diabetes-ss2_T2D-5.rds
Ignored: data/sce_iPSC-ss2_D0-joxm39.rds
Ignored: data/sce_iPSC-ss2_D0.rds
Ignored: output/00_Figures/
Ignored: output/01_sims/
Ignored: output/demo_eQTL/
Ignored: references/1000GP_Phase3_sample_info.txt
Ignored: references/Homo_sapiens.GRCh38.99.chromosome.22.gff3
Ignored: references/chr2.filt.2-temporary.bed
Ignored: references/chr2.filt.2-temporary.bim
Ignored: references/chr2.filt.2-temporary.fam
Ignored: references/chr2.filt.2.log
Ignored: references/chr2.filt.log
Ignored: references/chr2.filt.map
Ignored: references/chr2.filt.nosex
Ignored: references/chr2.filt.ped
Ignored: references/chr2.filt.prune.in
Ignored: references/chr2.filt.prune.out
Ignored: references/chr2.filtered.log
Ignored: references/chr2.filtered.nosex
Ignored: references/chr2.filtered.vcf
Ignored: references/chr2.filtered2.log
Ignored: references/chr2.filtered2.nosex
Ignored: references/chr2.filtered2.vcf
Ignored: references/chr2.filtered3.log
Ignored: references/chr2.filtered3.nosex
Ignored: references/chr2.filtered3.vcf
Ignored: references/chr2.genes.gff3
Ignored: references/chr2.vcf.gz
Ignored: references/chr22.filt.log
Ignored: references/chr22.filt.map
Ignored: references/chr22.filt.nosex
Ignored: references/chr22.filt.ped
Ignored: references/chr22.filt.prune.in
Ignored: references/chr22.filt.prune.out
Ignored: references/chr22.filtered.log
Ignored: references/chr22.filtered.nosex
Ignored: references/chr22.filtered.vcf
Ignored: references/chr22.filtered.vcf.bed
Ignored: references/chr22.filtered.vcf.bim
Ignored: references/chr22.filtered.vcf.fam
Ignored: references/chr22.filtered.vcf.log
Ignored: references/chr22.filtered.vcf.nosex
Ignored: references/chr22.filtered.vcf.rel
Ignored: references/chr22.filtered.vcf.rel.id
Ignored: references/chr22.filtered.vcf.rel_mod
Ignored: references/chr22.genes.gff3
Ignored: references/chr22.genes.gff3_annotation
Ignored: references/chr22.genes.gff3_chunks
Ignored: references/chr22.vcf.gz
Ignored: references/keep_samples.txt
Ignored: references/remove_snps.txt
Ignored: references/test.log
Ignored: references/test.nosex
Untracked files:
Untracked: .DS_Store
Untracked: .cache/
Untracked: .config/
Untracked: .snakemake/
Untracked: KEJP_2020_splatPop.Rproj
Untracked: Rplots.pdf
Untracked: analysis/10x-Fibrob_estimate-params.Rmd
Untracked: analysis/10x-Fibrob_simulations.Rmd
Untracked: analysis/10x-LungCarcinoma_estimate-params.Rmd
Untracked: analysis/splatPop_side-experiments.Rmd
Untracked: analysis/tmp_estimate-params.Rmd
Untracked: docs/
Untracked: mod
Untracked: raw_data.csv
Untracked: scRecover+scImpute.csv
Untracked: tempFile/
Untracked: test_data.rds
Untracked: test_unimputed.csv
Untracked: test_unimputed.rds
Unstaged changes:
Modified: .gitignore
Modified: .gitlab-ci.yml
Modified: CITATION
Modified: Dockerfile
Modified: LICENSE
Modified: README.md
Modified: _workflowr.yml
Modified: analysis/10x-Neuro_estimate-params.Rmd
Modified: analysis/KEJP_iPSC-ss2.Rmd
Modified: analysis/_site.yml
Modified: analysis/about.Rmd
Modified: analysis/index.Rmd
Modified: analysis/license.Rmd
Modified: analysis/ss2-iPSC_estimate-params.Rmd
Modified: analysis/ss2-iPSC_simulations.Rmd
Modified: cluster.json
Modified: code/1_process-empirical-data.R
Modified: code/2_estimate-splatPopParams.R
Modified: code/4_simulate_DEG.R
Modified: code/README.md
Modified: code/plot_functions.R
Modified: code/plot_functions2.R
Modified: data/README.md
Modified: environment.yml
Modified: envs/limix_env.yaml
Modified: envs/myenv.yaml
Modified: org/README.md
Modified: org/project_management.org
Modified: output/README.md
Modified: references/README.md
Modified: resources/README.md
Modified: resources/keep_samples.txt
Modified: workflow/config_bp_ss2.yaml
Modified: workflow/sims_limix_v1.15.1.smk
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
suppressPackageStartupMessages({
library(data.table)
library(SingleCellExperiment)
library(SingleR)
library(scPipe)
library(org.Mm.eg.db)
library(scater)
library(scran)
library(tidyverse)
library(Matrix)
library(splatter)
library(fitdistrplus)
library(RColorBrewer)
library(ggpubr)
})
source("code/plot_functions.R")
source("code/misc_functions.R")set.seed(42)
n.genes <- 504
save <- TRUE
rerun <- FALSE
sample.colors <- projectColors("samples")Process empirical data
The empirical data and the preprocessing performed is described in Peyser et al., 2018. The processed data is available for download at EBI. The cells were downloaded pre-normalized. Two fibrosis treatments were used, only the bleomycin 1.75 milligram per kilogram cells are included in this analysis.
if(rerun) {
# Convert to SCE object
counts <- readMM("data/lung_fibrosis/E-HCAD-14.aggregated_filtered_normalised_counts.mtx")
c.rows <- read.table("data/lung_fibrosis/E-HCAD-14.aggregated_filtered_normalised_counts.mtx_rows", sep="\t")
c.cols <- scan("data/lung_fibrosis/E-HCAD-14.aggregated_filtered_normalised_counts.mtx_cols", what="", sep="\n")
row.names(counts) <- c.rows$V1
colnames(counts) <- c.cols
meta <- read.csv("data/lung_fibrosis/ExpDesign-E-HCAD-14.tsv", sep="\t")
sce <- SingleCellExperiment(assays = list(counts = counts),
colData = meta, rowData = c.rows)
# Standardize col data names
colnames(colData(sce))[colnames(colData(sce)) == "Sample.Characteristic.disease."] <- "Condition"
colnames(colData(sce))[colnames(colData(sce)) == "Sample.Characteristic.individual."] <- "Sample"
sce$Batch <- "Batch1"
# Only keep control and bleomycin-induced fibrosis mice.
sce.subset <- subset(sce, , Factor.Value.compound. %in% c("none", "bleomycin 1.75 milligram per kilogram"))
sce.subset <- logNormCounts(sce.subset)
}According to the original analysis, fibroblast cells showed the biggest transcriptional difference between control and fibrosis samples. However, the downloaded data did not come with cell-type annotations. Here I will annotate cells automatically using SingleR. I also remove genes with duplicate symbols. The number of cells of each cell type and the number of fibroblast cells for each sample by condition are shown:
if (rerun) {
symbolID <- mapIds(org.Mm.eg.db, keys=rownames(sce.subset),
column="SYMBOL", keytype="ENSEMBL")
rowData(sce.subset)$Symbol <- symbolID
sce.subset <- sce.subset[!is.na(rowData(sce.subset)$Symbol), ]
rownames(sce.subset) <- rowData(sce.subset)$Symbol
ref <- celldex::MouseRNAseqData()
pred <- SingleR(test=sce.subset, ref=ref, labels=ref$label.main)
sce.subset$Group <- pred$labels
table(sce.subset$Group)
sce.subset2 <- subset(sce.subset, , Group == "Fibroblasts")
## keep all the not (!) duplicated genes
dupes <- duplicated(rownames(sce.subset2))
sce.subset2 <- sce.subset2[!dupes, ]
## Drop cells with >95% zeros
cellDropOut <- colSums(counts(sce.subset2) == 0) / nrow(sce.subset2)
cellsKeep <- names(which(cellDropOut <= 0.95))
sce.subset2 <- sce.subset2[, cellsKeep]
if(save){saveRDS(sce.subset2, file = "data/sce_10X-fibroblasts-allGenes.rds")}
} else {
sce.subset2 <- readRDS("data/sce_10X-fibroblasts-allGenes.rds")
}
table(sce.subset2$Sample, sce.subset2$Condition)
normal pulmonary fibrosis
947170 399 0
947172 174 0
947173 0 222
947174 0 221
947176 0 297
955736 349 0
955737 175 0
955738 0 373Gene selection
Gene selection was critical for this data set because it had relatively deep sequencing for a 10x experiment, but still had high levels of “dropout”. For example, even after removing cells with >95% zeros, there were many genes for which some cells would have counts in the high 100s, while others would be zero. This resulted in our estimated dispersion parameter being highly over-inflated (e.g. 10), leading to simulations with extreme cell outliers that were not observed in the empirical data. Thus we filtered to only keep genes with < 70% zeros, note that this also removed genes with a mean or variance of zero. We filtered genes using just cells from the control sample with the most cells (947170), as these are the cells that will be used to estimate single-cell splatter parameters.
Finally, the Peyser et al., 2018 paper highlighted genes they identified as differentially expressed between control and induced-fibrosis fibroblast cells. Because most genes aren’t differentially expressed, but we want to demonstrate the utility of splatPop for showing differences between conditions, we keep these known DEGs in our set of 504 genes. After filtering out genes with high dropout and keeping known DEGs, the remaining genes were randomly selected.
sample.use <- "947170"
# Remove outliers for gene mean (>99th) & genes with no variance.
t.count <- as.matrix(counts(subset(sce.subset2, , Sample %in% c(sample.use))))
geneMeans <- rowMeans(t.count)
geneDropOut <- rowSums(t.count == 0) / ncol(t.count)
genes.rm <- which(geneDropOut >= 0.5 | geneMeans >= quantile(geneMeans, 0.99))
# Known DE genes to keep
genes.DEG <- c("Col1a1", "Fn1", "Actg2", "Tagln", "Tpm2", "Des",
"Spp1", "Thbs1", "Tnc", "Timp1", "Col5a1", "Cthrc1", "Eln",
"Serpine1", "Postn", "Ltbp2", "Igfbp2")
genes.DEG <- setdiff(genes.DEG, genes.rm)
n.rand.genes <- n.genes - length(genes.DEG)
genes.random <- setdiff(rownames(sce.subset2), c(genes.DEG, names(genes.rm)))
genes.keep <- c(genes.DEG, sample(genes.random, n.rand.genes))
sce.subset3 <- sce.subset2[genes.keep, ]
if(save){ saveRDS(sce.subset3, file = "data/sce_10X-fibroblasts.rds") }Visualize empirical data
plotSims(sim=sce.subset3, variables = c("Sample", "Condition"), maxCells = 50,
colour_by = "Sample", shape_by= "Condition")Scale for 'colour' is already present. Adding another scale for 'colour',
which will replace the existing scale.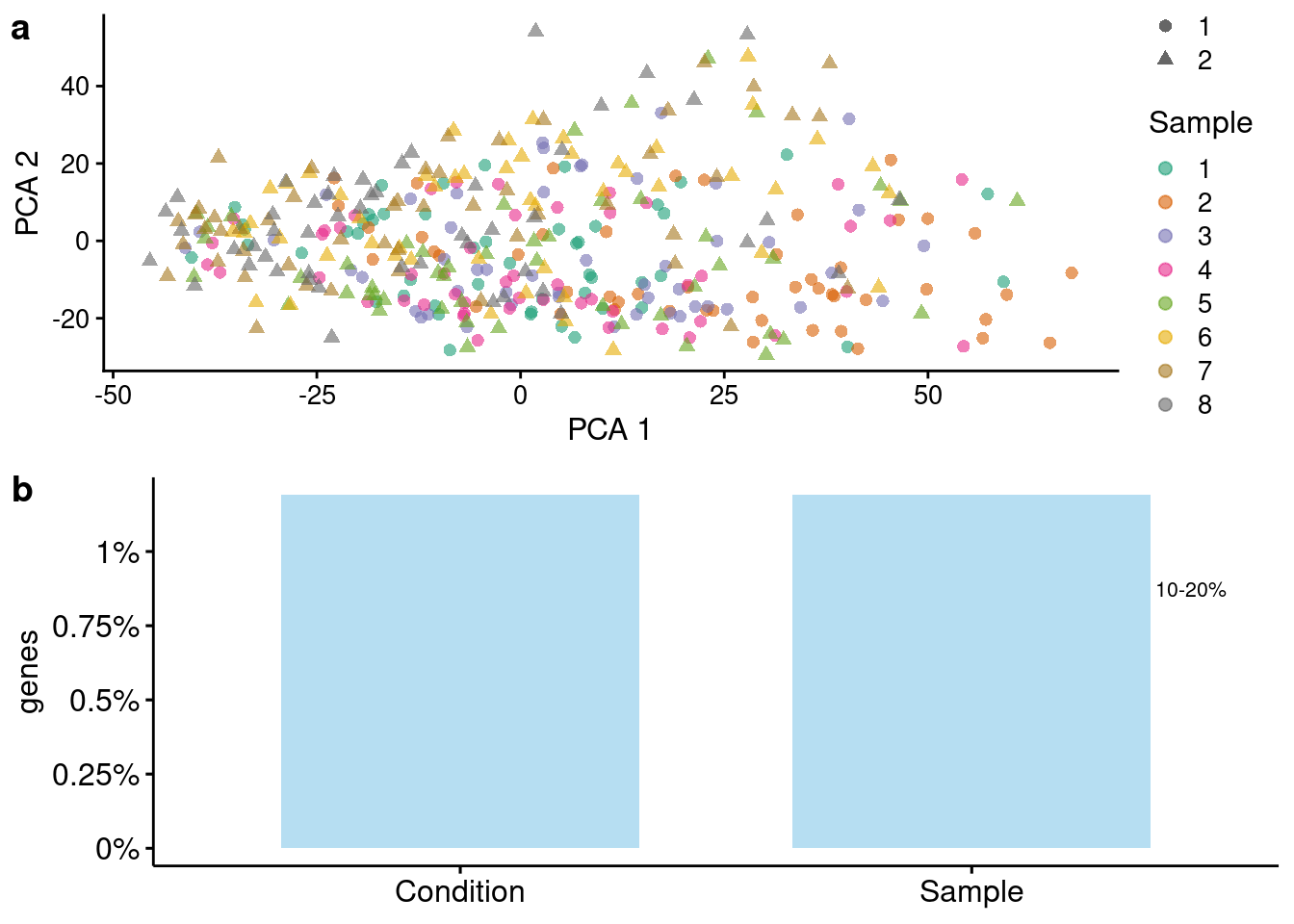
Generate mean aggregated data and single-cell SCE for the control sample with the most fibroblast cells (ID: 947170, n.cells: 399).
# Aggregate into population wide data
sce.control <- subset(sce.subset3, , Condition == "normal")
agg.10x <- aggregateAcrossCells(sce.control, ids = sce.control$Sample,
statistics="mean")
# Get cells from control sample with most cells
sce.single <- subset(sce.subset3, , Sample %in% c(sample.use))
if(save){
saveRDS(agg.10x, file = "data/agg_10X-fibroblasts-control.rds")
saveRDS(sce.single, file = "data/sce_10X-fibroblasts-947170.rds")
}Estimate splatPop parameters
Single-cell parameters are estimated from scRNA-seq data from cells from the donor with the most cells (947170) using 504 genes. Down-sampling the number of genes to match the number of genes we will simulate ensures the estimated library size parameters reflect real data.
Population parameters are estimated from either mean aggregated scRNA-seq data from non-diseased samples only. When mean aggregation is used we set pop.quant.norm to False because quantile normalization is not needed.
Parameters estimated for 10x fibroblasts:
params <- newSplatPopParams(pop.cv.bins = 20)
params <- splatPopEstimate(params = params,
counts = as.matrix(counts(sce.single)),
means = as.matrix(counts(agg.10x)))NOTE: Library sizes have been found to be normally distributed instead of log-normal. You may want to check this is correct.params <- setParams(params, pop.quant.norm = FALSE)
getParams(params, c("bcv.common", "bcv.df"))$bcv.common
[1] 1.302882
$bcv.df
[1] 31.32569if(save){
saveRDS(params, file = "output/01_sims/splatPop-params_10x-fibroblasts.rds")
}
paramsA Params object of class SplatPopParams
Parameters can be (estimable) or [not estimable], 'Default' or 'NOT DEFAULT'
Secondary parameters are usually set during simulation
Global:
(GENES) (CELLS) [Seed]
504 399 617511
53 additional parameters
Batches:
[BATCHES] [BATCH CELLS] [Location] [Scale] [Remove]
1 399 0.1 0.1 FALSE
Mean:
(RATE) (SHAPE)
0.0188917955612061 4.53603891347788
Library size:
(LOCATION) (SCALE) (NORM)
128388.066312588 10833.2945585279 TRUE
Exprs outliers:
(PROBABILITY) (LOCATION) (SCALE)
0.0277777777777778 1.1033453280682 0.0457624207463561
Groups:
[Groups] [Group Probs]
1 1
Diff expr:
[Probability] [Down Prob] [Location] [Scale]
0.1 0.5 0.1 0.4
BCV:
(COMMON DISP) (DOF)
1.30288198960497 31.3256891908181
Dropout:
[Type] (MIDPOINT) (SHAPE)
none 4.54331800813708 -0.979405502474792
Paths:
[From] [Steps] [Skew] [Non-linear] [Sigma Factor]
0 100 0.5 0.1 0.8
Population params:
(MEAN.SHAPE) (MEAN.RATE) [POP.QUANT.NORM] [similarity.scale]
3.2237556363283 0.0127349786312928 FALSE 1
[batch.size] [nCells.sample] [nCells.shape] [nCells.rate]
10 FALSE 1.5 0.015
[CV.BINS]
20
(CV.PARAMS)
data.frame (20 x 4) with columns: start, end, shape, rate
start end shape rate
1 0 103 3.809967 11.02002
2 103 119 5.049382 21.28337
3 119 129 3.072303 17.97121
4 129 139 5.726824 40.99890
# ... with 16 more rows
eQTL params:
[eqtl.n] [eqtl.dist]
0.5 1e+06
[eqtl.maf.min] [eqtl.maf.max]
0.05 0.5
[eqtl.group.specific] [eqtl.condition.specific]
0.2 0.2
(eqtl.ES.shape) (eqtl.ES.rate)
3.6 12
Condition params:
[nConditions] [condition.prob] [cde.prob] [cde.downProb]
1 1 0.1 0.5
[cde.facLoc] [cde.facScale]
0.1 0.4
devtools::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.0.4 (2021-02-15)
os Red Hat Enterprise Linux
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2021-06-02
─ Packages ───────────────────────────────────────────────────────────────────
package * version date lib source
abind 1.4-5 2016-07-21 [1] CRAN (R 4.0.2)
AnnotationDbi * 1.52.0 2020-10-27 [1] Bioconductor
askpass 1.1 2019-01-13 [1] CRAN (R 4.0.2)
assertthat 0.2.1 2019-03-21 [1] CRAN (R 4.0.2)
backports 1.2.1 2020-12-09 [1] CRAN (R 4.0.4)
beachmat 2.6.4 2020-12-20 [1] Bioconductor
beeswarm 0.3.1 2021-03-07 [1] CRAN (R 4.0.4)
Biobase * 2.50.0 2020-10-27 [1] Bioconductor
BiocFileCache 1.14.0 2020-10-27 [1] Bioconductor
BiocGenerics * 0.36.1 2021-04-16 [1] Bioconductor
BiocNeighbors 1.8.2 2020-12-07 [1] Bioconductor
BiocParallel 1.24.1 2020-11-06 [1] Bioconductor
BiocSingular 1.6.0 2020-10-27 [1] Bioconductor
biomaRt 2.46.3 2021-02-09 [1] Bioconductor
bit 4.0.4 2020-08-04 [1] CRAN (R 4.0.2)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.0.2)
bitops 1.0-7 2021-04-24 [1] CRAN (R 4.0.4)
blob 1.2.1 2020-01-20 [1] CRAN (R 4.0.2)
bluster 1.0.0 2020-10-27 [1] Bioconductor
broom 0.7.6 2021-04-05 [1] CRAN (R 4.0.4)
bslib 0.2.4 2021-01-25 [1] CRAN (R 4.0.3)
cachem 1.0.4 2021-02-13 [1] CRAN (R 4.0.3)
callr 3.7.0 2021-04-20 [1] CRAN (R 4.0.4)
car 3.0-10 2020-09-29 [1] CRAN (R 4.0.2)
carData 3.0-4 2020-05-22 [1] CRAN (R 4.0.2)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.0.2)
checkmate 2.0.0 2020-02-06 [1] CRAN (R 4.0.2)
cli 2.5.0 2021-04-26 [1] CRAN (R 4.0.4)
colorspace 2.0-1 2021-05-04 [1] CRAN (R 4.0.4)
cowplot 1.1.1 2020-12-30 [1] CRAN (R 4.0.4)
crayon 1.4.1 2021-02-08 [1] CRAN (R 4.0.4)
curl 4.3.1 2021-04-30 [1] CRAN (R 4.0.4)
data.table * 1.14.0 2021-02-21 [1] CRAN (R 4.0.4)
DBI 1.1.1 2021-01-15 [1] CRAN (R 4.0.4)
dbplyr 2.1.1 2021-04-06 [1] CRAN (R 4.0.4)
DelayedArray 0.16.0 2020-10-27 [1] Bioconductor
DelayedMatrixStats 1.12.3 2021-02-03 [1] Bioconductor
DEoptimR 1.0-9 2021-05-24 [1] CRAN (R 4.0.4)
desc 1.3.0 2021-03-05 [1] CRAN (R 4.0.4)
devtools 2.3.2 2020-09-18 [1] CRAN (R 4.0.2)
digest 0.6.27 2020-10-24 [1] CRAN (R 4.0.2)
dplyr * 1.0.5 2021-03-05 [1] CRAN (R 4.0.4)
dqrng 0.3.0 2021-05-01 [1] CRAN (R 4.0.4)
edgeR 3.32.1 2021-01-14 [1] Bioconductor
ellipsis 0.3.1 2020-05-15 [1] CRAN (R 4.0.2)
evaluate 0.14 2019-05-28 [1] CRAN (R 4.0.2)
fansi 0.4.2 2021-01-15 [1] CRAN (R 4.0.4)
farver 2.1.0 2021-02-28 [1] CRAN (R 4.0.4)
fastmap 1.1.0 2021-01-25 [1] CRAN (R 4.0.3)
fitdistrplus * 1.1-3 2020-12-05 [1] CRAN (R 4.0.3)
forcats * 0.5.1 2021-01-27 [1] CRAN (R 4.0.4)
foreign 0.8-81 2020-12-22 [2] CRAN (R 4.0.4)
fs 1.5.0 2020-07-31 [1] CRAN (R 4.0.2)
generics 0.1.0 2020-10-31 [1] CRAN (R 4.0.2)
GenomeInfoDb * 1.26.7 2021-04-08 [1] Bioconductor
GenomeInfoDbData 1.2.4 2020-11-10 [1] Bioconductor
GenomicRanges * 1.42.0 2020-10-27 [1] Bioconductor
GGally 2.1.1 2021-03-08 [1] CRAN (R 4.0.4)
ggbeeswarm 0.6.0 2017-08-07 [1] CRAN (R 4.0.2)
ggplot2 * 3.3.3 2020-12-30 [1] CRAN (R 4.0.4)
ggpubr * 0.4.0 2020-06-27 [1] CRAN (R 4.0.3)
ggsignif 0.6.1 2021-02-23 [1] CRAN (R 4.0.3)
git2r 0.28.0 2021-01-10 [1] CRAN (R 4.0.4)
glue 1.4.2 2020-08-27 [1] CRAN (R 4.0.2)
gridExtra 2.3 2017-09-09 [1] CRAN (R 4.0.2)
gtable 0.3.0 2019-03-25 [1] CRAN (R 4.0.2)
haven 2.4.0 2021-04-14 [1] CRAN (R 4.0.4)
highr 0.9 2021-04-16 [1] CRAN (R 4.0.4)
hms 1.0.0 2021-01-13 [1] CRAN (R 4.0.4)
htmltools 0.5.1.1 2021-01-22 [1] CRAN (R 4.0.3)
httpuv 1.5.5 2021-01-13 [1] CRAN (R 4.0.4)
httr 1.4.2 2020-07-20 [1] CRAN (R 4.0.2)
igraph 1.2.6 2020-10-06 [1] CRAN (R 4.0.2)
IRanges * 2.24.0 2020-10-27 [1] Bioconductor
irlba 2.3.3 2019-02-05 [1] CRAN (R 4.0.2)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.0.4)
jsonlite 1.7.2 2020-12-09 [1] CRAN (R 4.0.4)
knitr 1.32 2021-04-14 [1] CRAN (R 4.0.4)
labeling 0.4.2 2020-10-20 [1] CRAN (R 4.0.2)
later 1.1.0.1 2020-06-05 [1] CRAN (R 4.0.2)
lattice 0.20-41 2020-04-02 [2] CRAN (R 4.0.4)
lifecycle 1.0.0 2021-02-15 [1] CRAN (R 4.0.4)
limma 3.46.0 2020-10-27 [1] Bioconductor
locfit 1.5-9.4 2020-03-25 [1] CRAN (R 4.0.2)
lubridate 1.7.10 2021-02-26 [1] CRAN (R 4.0.4)
magrittr 2.0.1 2020-11-17 [1] CRAN (R 4.0.3)
MASS * 7.3-54 2021-05-03 [1] CRAN (R 4.0.4)
Matrix * 1.3-3 2021-05-04 [1] CRAN (R 4.0.4)
MatrixGenerics * 1.2.0 2020-10-27 [1] Bioconductor
matrixStats * 0.58.0 2021-01-29 [1] CRAN (R 4.0.4)
mclust 5.4.7 2020-11-20 [1] CRAN (R 4.0.3)
memoise 2.0.0 2021-01-26 [1] CRAN (R 4.0.4)
modelr 0.1.8 2020-05-19 [1] CRAN (R 4.0.2)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.0.2)
openssl 1.4.4 2021-04-30 [1] CRAN (R 4.0.4)
openxlsx 4.2.3 2020-10-27 [1] CRAN (R 4.0.2)
org.Hs.eg.db 3.12.0 2021-04-28 [1] Bioconductor
org.Mm.eg.db * 3.12.0 2021-05-18 [1] Bioconductor
pillar 1.6.0 2021-04-13 [1] CRAN (R 4.0.4)
pkgbuild 1.2.0 2020-12-15 [1] CRAN (R 4.0.4)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.0.2)
pkgload 1.2.1 2021-04-06 [1] CRAN (R 4.0.4)
plyr 1.8.6 2020-03-03 [1] CRAN (R 4.0.2)
prettyunits 1.1.1 2020-01-24 [1] CRAN (R 4.0.2)
processx 3.5.2 2021-04-30 [1] CRAN (R 4.0.4)
progress 1.2.2 2019-05-16 [1] CRAN (R 4.0.2)
promises 1.2.0.1 2021-02-11 [1] CRAN (R 4.0.4)
ps 1.6.0 2021-02-28 [1] CRAN (R 4.0.4)
purrr * 0.3.4 2020-04-17 [1] CRAN (R 4.0.2)
R6 2.5.0 2020-10-28 [1] CRAN (R 4.0.2)
rappdirs 0.3.3 2021-01-31 [1] CRAN (R 4.0.4)
RColorBrewer * 1.1-2 2014-12-07 [1] CRAN (R 4.0.2)
Rcpp 1.0.6 2021-01-15 [1] CRAN (R 4.0.4)
RCurl 1.98-1.3 2021-03-16 [1] CRAN (R 4.0.4)
readr * 1.4.0 2020-10-05 [1] CRAN (R 4.0.2)
readxl 1.3.1 2019-03-13 [1] CRAN (R 4.0.2)
remotes 2.3.0 2021-04-01 [1] CRAN (R 4.0.4)
reprex 2.0.0 2021-04-02 [1] CRAN (R 4.0.4)
reshape 0.8.8 2018-10-23 [1] CRAN (R 4.0.4)
Rhtslib 1.22.0 2020-10-27 [1] Bioconductor
rio 0.5.26 2021-03-01 [1] CRAN (R 4.0.4)
rlang 0.4.10 2020-12-30 [1] CRAN (R 4.0.4)
rmarkdown 2.7 2021-02-19 [1] CRAN (R 4.0.4)
robustbase 0.93-7 2021-01-04 [1] CRAN (R 4.0.4)
rprojroot 2.0.2 2020-11-15 [1] CRAN (R 4.0.3)
RSQLite 2.2.1 2020-09-30 [1] CRAN (R 4.0.2)
rstatix 0.7.0 2021-02-13 [1] CRAN (R 4.0.4)
rstudioapi 0.13 2020-11-12 [1] CRAN (R 4.0.3)
rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.0.4)
rvest 1.0.0 2021-03-09 [1] CRAN (R 4.0.4)
S4Vectors * 0.28.0 2020-10-27 [1] Bioconductor
sass 0.3.1 2021-01-24 [1] CRAN (R 4.0.3)
scales 1.1.1 2020-05-11 [1] CRAN (R 4.0.2)
scater * 1.18.6 2021-02-26 [1] Bioconductor
scPipe * 1.12.0 2020-10-27 [1] Bioconductor
scran * 1.18.7 2021-04-16 [1] Bioconductor
scuttle 1.0.3 2020-11-23 [1] Bioconductor
sessioninfo 1.1.1 2018-11-05 [1] CRAN (R 4.0.2)
SingleCellExperiment * 1.12.0 2020-10-27 [1] Bioconductor
SingleR * 1.4.1 2021-02-02 [1] Bioconductor
sparseMatrixStats 1.2.1 2021-02-02 [1] Bioconductor
splatter * 1.15.2 2021-04-15 [1] Bioconductor
statmod 1.4.35 2020-10-19 [1] CRAN (R 4.0.2)
stringi 1.5.3 2020-09-09 [1] CRAN (R 4.0.2)
stringr * 1.4.0 2019-02-10 [1] CRAN (R 4.0.2)
SummarizedExperiment * 1.20.0 2020-10-27 [1] Bioconductor
survival * 3.2-10 2021-03-16 [1] CRAN (R 4.0.4)
testthat 3.0.0 2020-10-31 [1] CRAN (R 4.0.2)
tibble * 3.1.1 2021-04-18 [1] CRAN (R 4.0.4)
tidyr * 1.1.3 2021-03-03 [1] CRAN (R 4.0.4)
tidyselect 1.1.0 2020-05-11 [1] CRAN (R 4.0.2)
tidyverse * 1.3.1 2021-04-15 [1] CRAN (R 4.0.4)
usethis 1.6.3 2020-09-17 [1] CRAN (R 4.0.2)
utf8 1.2.1 2021-03-12 [1] CRAN (R 4.0.4)
vctrs 0.3.7 2021-03-29 [1] CRAN (R 4.0.4)
vipor 0.4.5 2017-03-22 [1] CRAN (R 4.0.2)
viridis 0.6.0 2021-04-15 [1] CRAN (R 4.0.4)
viridisLite 0.4.0 2021-04-13 [1] CRAN (R 4.0.4)
withr 2.4.2 2021-04-18 [1] CRAN (R 4.0.4)
workflowr 1.6.2 2020-04-30 [1] CRAN (R 4.0.2)
xfun 0.22 2021-03-11 [1] CRAN (R 4.0.4)
XML 3.99-0.6 2021-03-16 [1] CRAN (R 4.0.4)
xml2 1.3.2 2020-04-23 [1] CRAN (R 4.0.2)
XVector 0.30.0 2020-10-27 [1] Bioconductor
yaml 2.2.1 2020-02-01 [1] CRAN (R 4.0.2)
zip 2.1.1 2020-08-27 [1] CRAN (R 4.0.2)
zlibbioc 1.36.0 2020-10-27 [1] Bioconductor
[1] /mnt/mcfiles/cazodi/R/x86_64-pc-linux-gnu-library/4.0
[2] /opt/R/4.0.4/lib/R/library