Method concordance
Jeffrey Pullin
01/01/24
Last updated: 2024-01-01
Checks: 7 0
Knit directory:
mage_2020_marker-gene-benchmarking/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20190102) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 2632193. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Renviron
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .snakemake/
Ignored: NSForest/.Rhistory
Ignored: NSForest/NS-Forest_v3_Extended_Binary_Markers_Supplmental.csv
Ignored: NSForest/NS-Forest_v3_Full_Results.csv
Ignored: NSForest/NSForest3_medianValues.csv
Ignored: NSForest/NSForest_v3_Final_Result.csv
Ignored: NSForest/__pycache__/
Ignored: NSForest/data/
Ignored: RankCorr/picturedRocks/__pycache__/
Ignored: benchmarks/
Ignored: config/
Ignored: data/cellmarker/
Ignored: data/downloaded_data/
Ignored: data/expert_annotations/
Ignored: data/expert_mgs/
Ignored: data/raw_data/
Ignored: data/real_data/
Ignored: data/sim_data/
Ignored: data/sim_mgs/
Ignored: data/special_real_data/
Ignored: figures/
Ignored: logs/
Ignored: results/
Ignored: weights/
Unstaged changes:
Deleted: analysis/expert-mgs-direction.Rmd
Modified: smash-fork
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/concordance.Rmd) and HTML
(public/concordance.html) files. If you’ve configured a
remote Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2632193 | Jeffrey Pullin | 2024-01-01 | Implement revisions |
| Rmd | bc98f8b | Jeffrey Pullin | 2023-11-20 | GB resubmission July 2023 |
| Rmd | f54229a | Jeffrey Pullin | 2023-06-17 | Draft additional concordance analysis |
| html | fcecf65 | Jeffrey Pullin | 2022-09-09 | Build site. |
| Rmd | 0c2eafc | Jeffrey Pullin | 2022-09-09 | Update website |
| html | af96b34 | Jeffrey Pullin | 2022-08-30 | Build site. |
| Rmd | 854d178 | Jeffrey Pullin | 2022-07-21 | Update concordance analysis |
| html | 0e47874 | Jeffrey Pullin | 2022-05-04 | Build site. |
| html | b5045c1 | Jeffrey Pullin | 2022-05-02 | Build site. |
| Rmd | 048156f | Jeffrey Pullin | 2022-05-02 | Tweak analysis outputs |
| html | 048156f | Jeffrey Pullin | 2022-05-02 | Tweak analysis outputs |
| html | 8b989e1 | Jeffrey Pullin | 2022-05-02 | Build site. |
| html | 0548273 | Jeffrey Pullin | 2022-05-02 | Build site. |
| Rmd | 50bca7c | Jeffrey Pullin | 2022-05-02 | workflowr::wflow_publish(all = TRUE, republish = TRUE) |
| html | 50bca7c | Jeffrey Pullin | 2022-05-02 | workflowr::wflow_publish(all = TRUE, republish = TRUE) |
| html | 5cc008f | Jeffrey Pullin | 2022-02-09 | Build site. |
| Rmd | d1aca16 | Jeffrey Pullin | 2022-02-09 | Refresh website |
| html | d1aca16 | Jeffrey Pullin | 2022-02-09 | Refresh website |
| Rmd | aca9ad2 | Jeffrey Pullin | 2021-11-29 | Various changes made in the last days before thesis submission |
| Rmd | d3804d1 | Jeffrey Pullin | 2021-09-23 | Polish analyses |
| Rmd | b520587 | Jeffrey Pullin | 2021-09-21 | Polish concordance analysis |
| Rmd | 211b2f6 | Jeffrey Pullin | 2021-09-21 | Add ranking plots concordance analysis |
| Rmd | 92d3bf0 | Jeffrey Pullin | 2021-08-02 | Add code to create plots for ECSSC talk |
| html | 61ee246 | Jeffrey Pullin | 2021-04-13 | Build site. |
| Rmd | b5b2a88 | Jeffrey Pullin | 2021-04-13 | Add new results |
| Rmd | b895f6e | Jeffrey Pullin | 2021-04-05 | Add cluster analysis |
| Rmd | d036935 | Jeffrey Pullin | 2021-03-31 | Refactor concordance analysis |
| html | ca82ce0 | Jeffrey Pullin | 2021-02-16 | Build site. |
| html | 2863555 | Jeffrey Pullin | 2021-02-10 | Build site. |
| Rmd | 1ad9d6d | Jeffrey Pullin | 2021-02-10 | Add workflowr website |
library(tibble)
library(dplyr)
library(ggupset)
library(ggplot2)
library(tidyr)
library(SingleCellExperiment)
library(logisticPCA)
library(pals)
library(topconfects)
library(patchwork)
library(ggrepel)
library(purrr)
library(scater)
library(forcats)
library(ggdendro)source(here::here("code", "top-genes.R"))
source(here::here("code", "analysis-utils.R"))
source(here::here("code", "plot-utils.R"))
#' Get the top marker genes for a specific cluster
#'
#' @param output The output of a (data, method) pair run
#' @param cluster The cluster to extract marker genes for
#' @param n The number of marker genes to extract
#'
#' @return A vector of gene names
#'
get_top_marker_genes <- function(output, cluster, n) {
result <- output$result
result <- result[result$cluster == cluster, ]
result <- get_top_sel_mgs(result, n = n)
mgs <- result$gene
mgs
}concordance_data <- retrieve_real_data_parameters() %>%
filter(!(data_id %in% c("citeseq", "mesenchymal", "ss3_pbmc", "zhao"))) %>%
rowwise() %>%
mutate(mgs = list(readRDS(full_filename)$result)) %>%
mutate(mgs = list(split(mgs, mgs$cluster))) %>%
ungroup() %>%
unnest(cols = mgs) %>%
rowwise() %>%
mutate(cluster = mgs$cluster[[1]]) %>%
ungroup()
poor_methods <- c("random", "binom", "difference", "lm")long_data <- concordance_data %>%
filter(!(method %in% poor_methods)) %>%
filter(data_id == "pbmc3k", cluster == "B") %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = 100)$gene)) %>%
ungroup() %>%
unnest_longer(col = mgs) %>%
# Not sure why this occurs.
filter(!is.na(mgs))
binary_data <- model.matrix(~ 0 + . , data = long_data["mgs"])
cluster_data <- cbind(
pars = long_data$pars,
as.data.frame(binary_data)
) %>%
group_by(pars) %>%
summarise(across(everything(), sum))
cluster_mat <- as.matrix(cluster_data[, -1])Dimensionality reduction: PCA
pca <- prcomp(cluster_mat)
pca_plot <- tibble(
pc1 = pca$x[, 1],
pc2 = pca$x[, 2],
pars = cluster_data$pars,
plot_pars = pars_lookup[cluster_data$pars]
) %>%
rowwise() %>%
mutate(method = strsplit(pars, "[_]")[[1]][1]) %>%
ungroup() %>%
ggplot(aes(x = pc1, y = pc2, label = plot_pars, colour = method)) +
geom_point(size = 3) +
geom_text_repel(
aes(label = plot_pars),
colour = "black",
max.overlaps = 20) +
labs(
x = "Principal component 1",
y = "Principal component 2",
colour = "Package"
) +
theme_bw()
pca_plot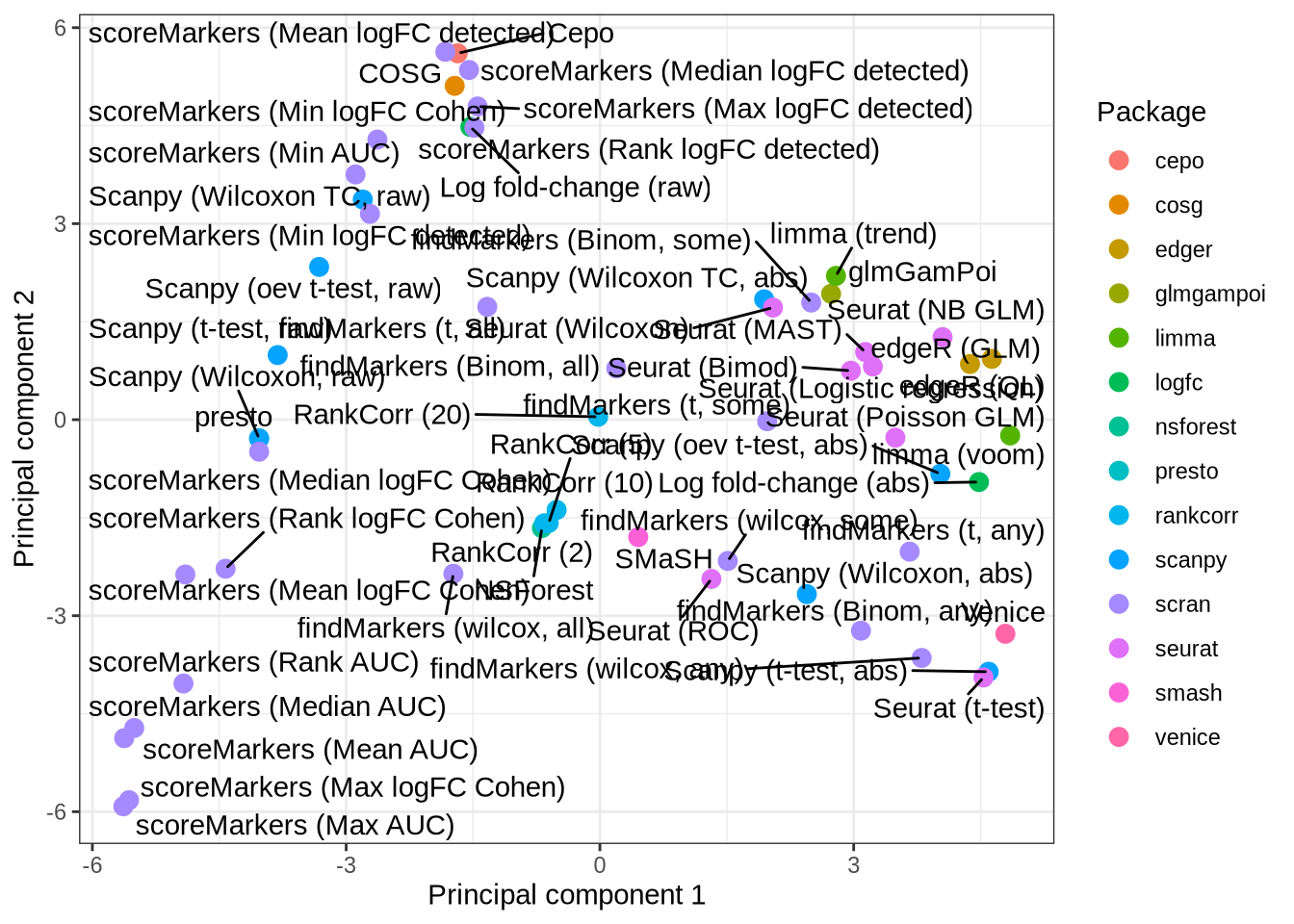
saveRDS(
pca_plot,
here::here("figures", "raw", "concordance-pca-plot-with-text.rds")
)Intersection dendrogram
n <- 10
all_concordance_data <- concordance_data %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = n)$gene)) %>%
ungroup() %>%
select(pars, method, cluster, data_id, mgs)
intersection_data <- all_concordance_data %>%
expand_grid(pars_2 = unique(all_concordance_data$pars)) %>%
left_join(
dplyr::rename(all_concordance_data, mgs_2 = mgs),
by = c("pars_2" = "pars", "cluster")
) %>%
rowwise() %>%
mutate(prop_intersect = length(intersect(mgs, mgs_2)) / n) %>%
ungroup() %>%
select(pars, pars_2, cluster, prop_intersect) %>%
group_by(pars, pars_2) %>%
summarise(prop_intersect = mean(prop_intersect), .groups = "drop") %>%
pivot_wider(names_from = pars, values_from = prop_intersect)
intersection_mat <- as.matrix(intersection_data[, -1])
rownames(intersection_mat) <- intersection_data[[1]]
hier_clust <- hclust(dist(intersection_mat))
hier_clust$labels <- pars_lookup[hier_clust$labels]
hier_clust_data <- dendro_data(hier_clust, type = "rectangle")
dendrogram_plot <- ggplot() +
geom_segment(data = segment(hier_clust_data),
aes(x = x, y = y, xend = xend, yend = yend)
) +
geom_text(data = label(hier_clust_data),
aes(x = x, y = y, label = label, hjust = 0),
size = 4
) +
coord_flip(xlim = c(3, 57)) +
scale_y_reverse(limits = c(4, -1.5)) +
theme_dendro()
dendrogram_plot
saveRDS(dendrogram_plot, here::here("figures", "raw", "dendrogram.rds"))raw_pars <- c("scanpy_tover_rankby_raw", "scanpy_wilcoxon_rankby_raw", "presto",
"logfc_raw", "scanpy_t_rankby_raw",
"scanpy_wilcoxontiecorrect_rankby_raw")
non_rank_pars <- c("rankcorr_2", "rankcorr_10", "rankcorr_20", "rankcorr_5",
"nsforest")
dendro_info_data <- tibble(
pars = rownames(hier_clust_data$labels),
) %>%
mutate(plot_pars = pars_lookup[pars]) %>%
mutate(plot_pars = factor(plot_pars, levels = hier_clust_data$labels$label)) %>%
mutate(
is_raw = as.character(pars %in% raw_pars),
is_non_rank = as.character(pars %in% non_rank_pars)
) %>%
left_join(
select(concordance_data, c(pars, method)),
by = "pars"
) %>%
mutate(plot_method = method_lookup[method]) %>%
pivot_longer(cols = c(is_raw, is_non_rank, plot_method),
names_to = "criteria")
is_raw_plot <- dendro_info_data %>%
filter(criteria == "is_raw") %>%
mutate(Direction = value) %>%
mutate(Direction = if_else(Direction == "FALSE",
"Either", "Up-regulated only")) %>%
mutate(Direction = factor(Direction, levels = c("Up-regulated only", "Either"))) %>%
ggplot(aes(x = criteria, y = plot_pars)) +
geom_tile(aes(fill = Direction), colour = "black") +
scale_fill_manual(values = c("#E41A1C", "#377EB8")) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
axis.title = element_blank()
)
is_raw_plot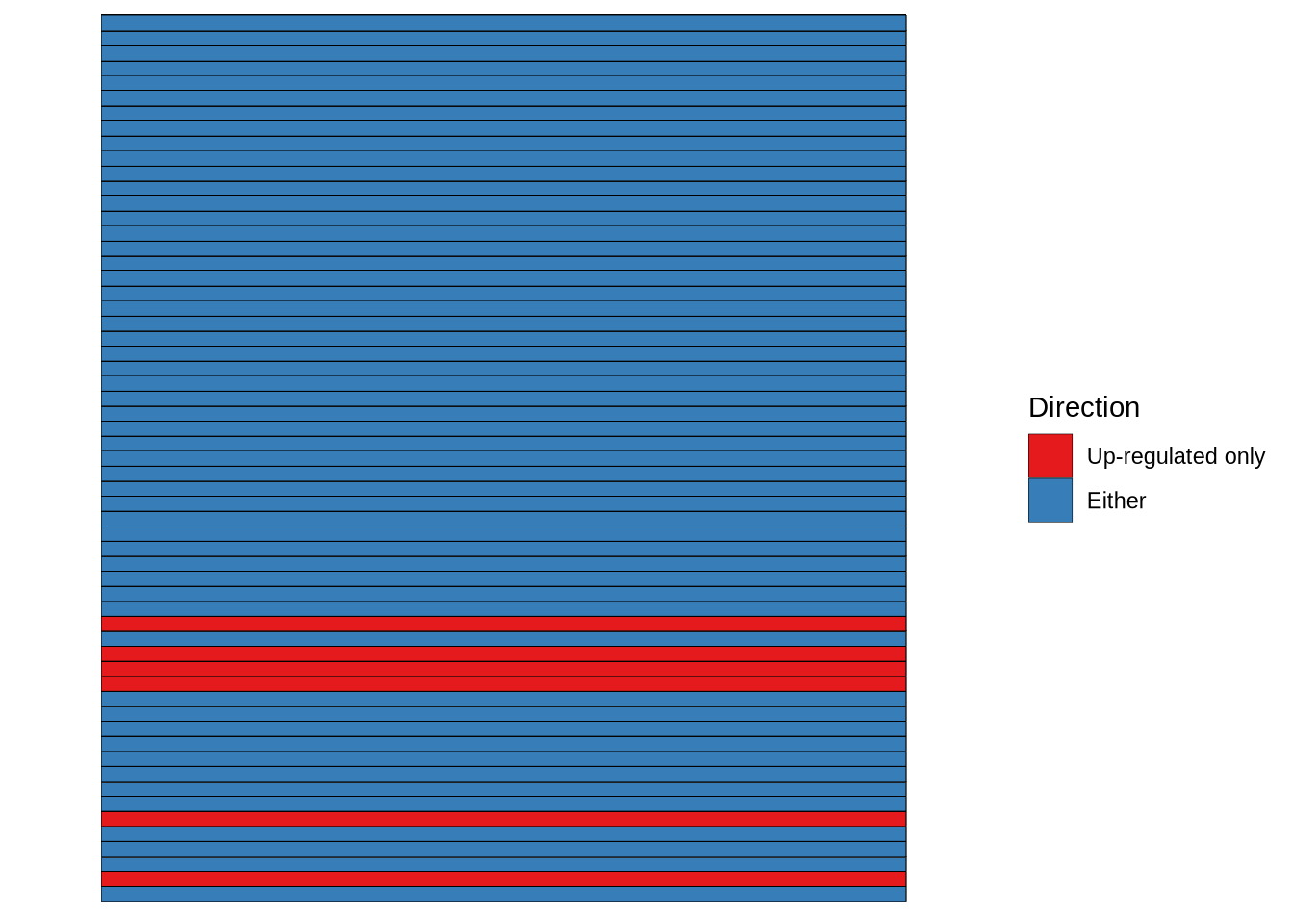
is_non_rank_plot <- dendro_info_data %>%
filter(criteria == "is_non_rank") %>%
mutate(Strategy = if_else(value == "FALSE",
"Ranking", "Fixed set")) %>%
mutate(`Rank or set` = factor(Strategy, levels = c("Fixed set", "Ranking"))) %>%
ggplot(aes(x = criteria, y = plot_pars)) +
geom_tile(aes(fill = `Rank or set`), colour = "black") +
scale_fill_manual(values = c("#66C2A5", "#FC8D62")) +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
axis.title = element_blank()
)
is_non_rank_plot
package_plot <- dendro_info_data %>%
filter(criteria == "plot_method") %>%
mutate(Package = value) %>%
ggplot(aes(x = criteria, y = plot_pars)) +
geom_tile(aes(fill = Package), colour = "black") +
package_fill +
theme_bw() +
theme(
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_blank(),
axis.text = element_blank(),
axis.ticks = element_blank(),
axis.title = element_blank()
)
package_plot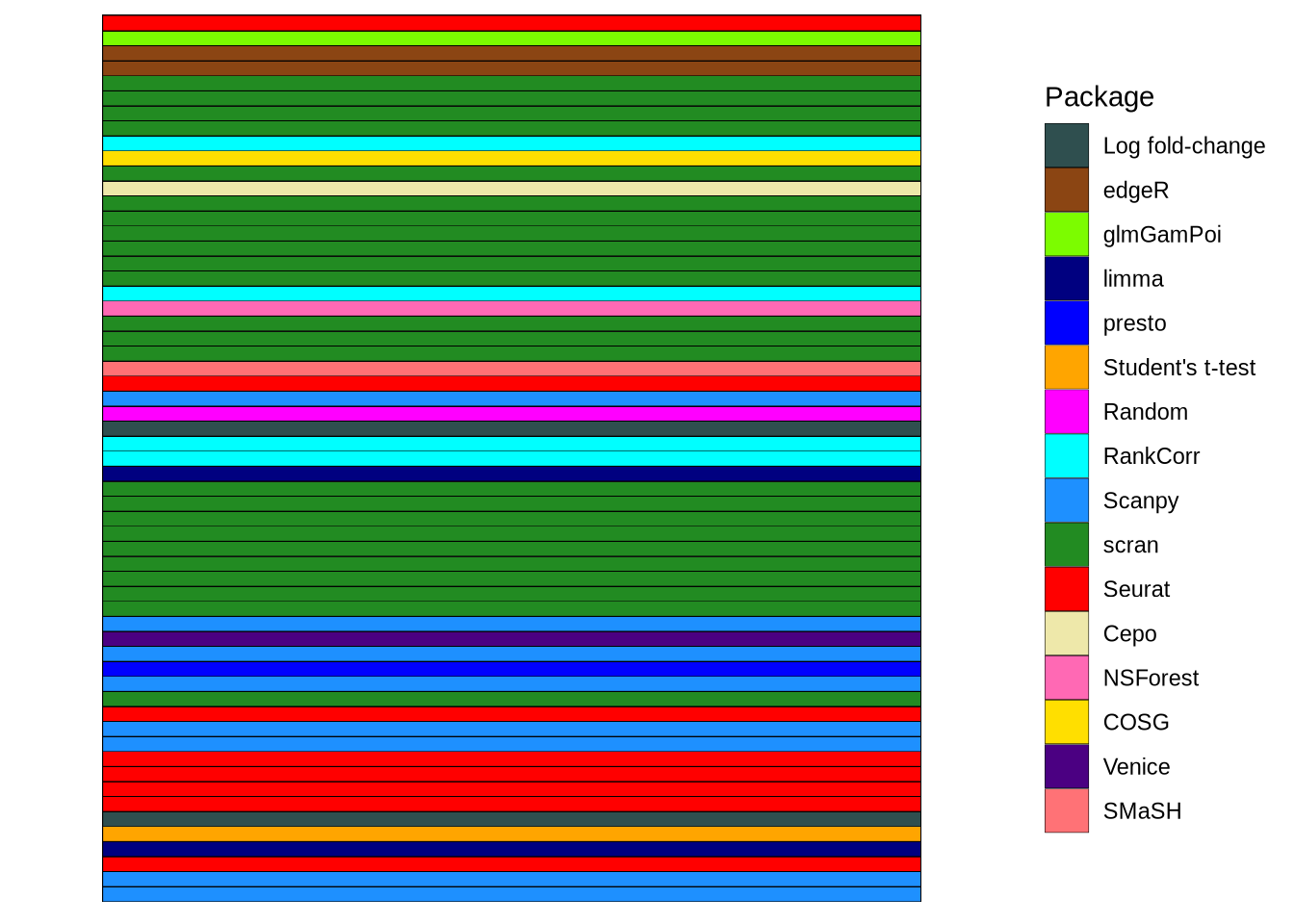
saveRDS(is_raw_plot, here::here("figures", "raw", "raw-info.rds"))
saveRDS(is_non_rank_plot, here::here("figures", "raw", "strategy-info.rds"))
saveRDS(package_plot, here::here("figures", "raw", "package-info.rds"))Dimensionality reduction: logistic PCA
Perform Logistic PCA
logistic_pca <- logisticPCA(cluster_mat)
logistic_pca_data <- tibble(
pc1 = logistic_pca$PCs[, 1],
pc2 = logistic_pca$PCs[, 2],
pars = cluster_data$pars,
plot_pars = pars_lookup[cluster_data$pars]
) %>%
rowwise() %>%
mutate(method = strsplit(pars, "[_]")[[1]][1]) %>%
ungroup()Plot logistic
ggplot(logistic_pca_data,
aes(x = pc1, y = pc2, label = plot_pars, colour = method)) +
geom_point(size = 3) +
geom_text_repel(
aes(label = plot_pars),
colour = "black",
max.overlaps = 20) +
labs(
x = "PC 1",
y = "PC 2",
colour = "Method"
) +
theme_bw()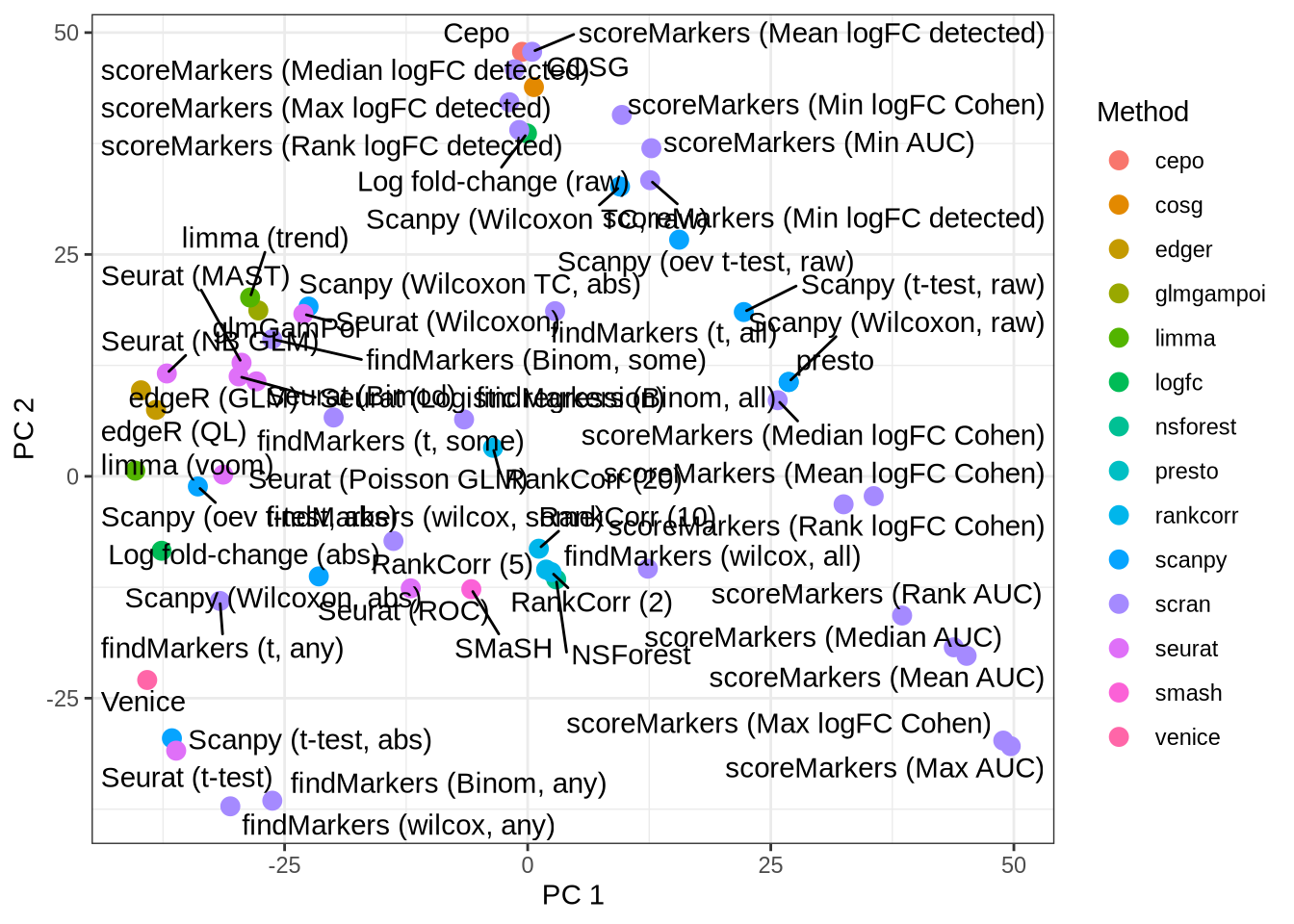
Intersection concordance analysis
all_datasets_results <- load_real_data_results("all")
n_genes <- 20
seurat_results <- all_datasets_results %>%
filter(pars == "seurat_wilcox") %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = n_genes))) %>%
ungroup() %>%
select(cluster, seurat_mgs = mgs)
scanpy_results <- all_datasets_results %>%
filter(pars == "scanpy_t_rankby_raw") %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = n_genes))) %>%
ungroup() %>%
select(cluster, scanpy_mgs = mgs, data_id)
scanpy_seurat_intersection_plot <- seurat_results %>%
left_join(scanpy_results, by = "cluster") %>%
rowwise() %>%
mutate(
prop_intersect = length(intersect(seurat_mgs$gene, scanpy_mgs$gene)) / n_genes
) %>%
ungroup() %>%
mutate(plot_data_id = dataset_lookup[data_id]) %>%
mutate(plot_data_id = fct_reorder(factor(plot_data_id), prop_intersect)) %>%
ggplot(aes(x = plot_data_id, y = prop_intersect)) +
geom_boxplot(outlier.shape = NA) +
geom_jitter(size = 2, width = 0.1, colour = "forestgreen") +
coord_flip(ylim = c(0, 1)) +
labs(
x = "Dataset",
y = "Proportion of shared genes"
) +
theme_bw()
scanpy_seurat_intersection_plot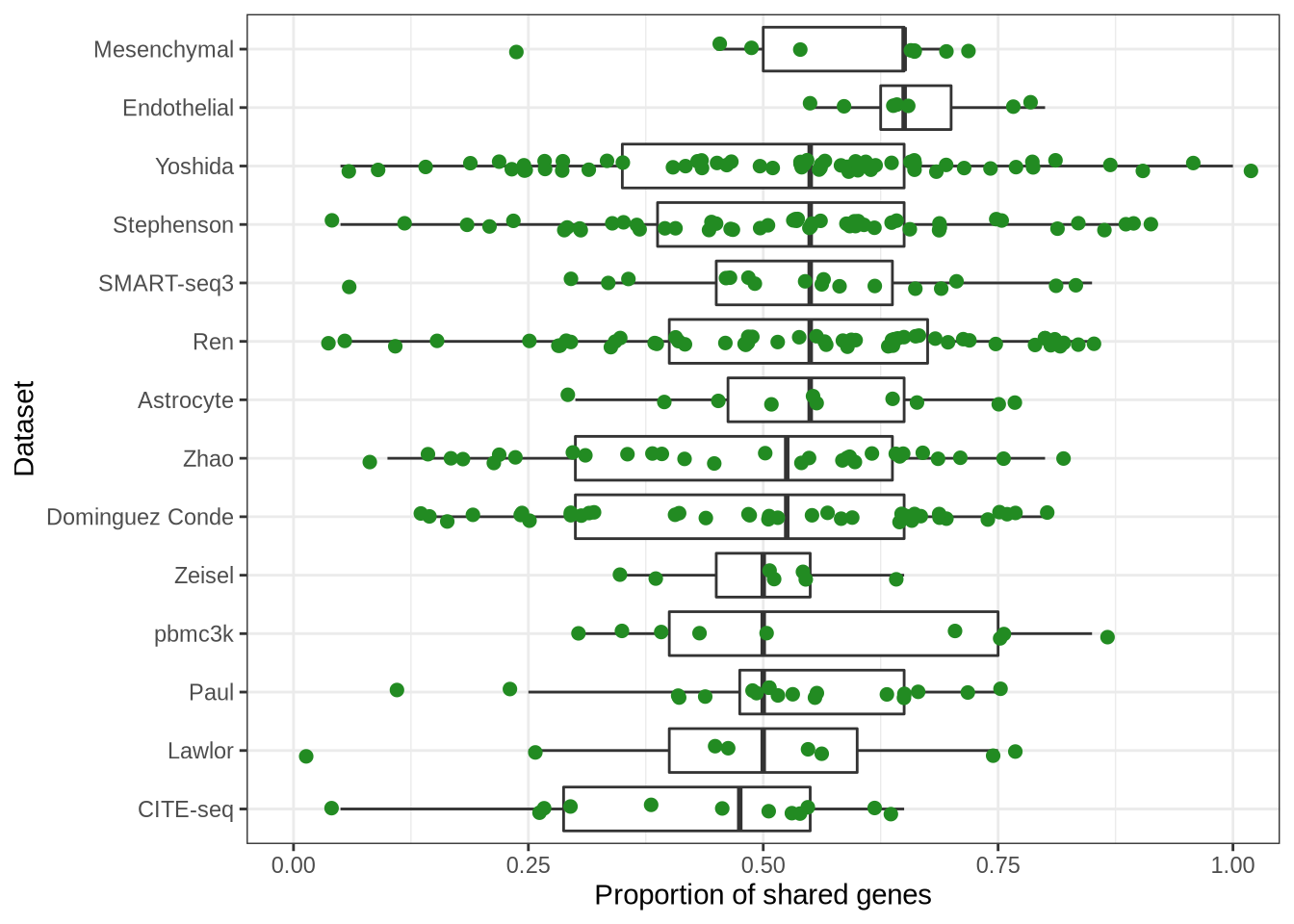
saveRDS(
scanpy_seurat_intersection_plot,
here::here("figures", "raw", "scanpy-seurat-intersection.rds")
)Visual rank concordance analysis
Unless otherwise stated these plots are generated for the B cell cluster in the pbmc3k dataset.
scanpy_t_raw <- readRDS(
here::here("results", "real_data", "pbmc3k-scanpy_t_rankby_raw.rds")
)
scanpy_t_abs <- readRDS(
here::here("results", "real_data", "pbmc3k-scanpy_t_rankby_abs.rds")
)
seurat_t <- readRDS(
here::here("results", "real_data", "pbmc3k-seurat_t.rds")
)
scanpy_wilcox <- readRDS(
here::here("results", "real_data", "pbmc3k-scanpy_wilcoxon_rankby_raw.rds")
)
scanpy_wilcox_tc <- readRDS(
here::here("results", "real_data",
"pbmc3k-scanpy_wilcoxontiecorrect_rankby_raw.rds")
)
scanpy_wilcox_tc_rankby_abs <- readRDS(
here::here("results", "real_data",
"pbmc3k-scanpy_wilcoxontiecorrect_rankby_abs.rds")
)
seurat_wilcox <- readRDS(
here::here("results", "real_data", "pbmc3k-seurat_wilcox.rds")
)
scran_t_any <- readRDS(
here::here("results", "real_data", "pbmc3k-scran_findMarkers_t_any.rds")
)Seurat default vs Scanpy default
scanpy_default_seurat_default_plot <- rank_rank_plot(
get_top_marker_genes(scanpy_t_raw, cluster = "CD8 T", n = 20),
get_top_marker_genes(seurat_wilcox, cluster = "CD8 T", n = 20),
label1 = "Scanpy default",
label2 = "Seurat default"
) +
ggtitle("Scanpy vs Seurat (CD8 T)") +
theme(axis.ticks.y = element_blank())
scanpy_default_seurat_default_plot![]()
saveRDS(scanpy_default_seurat_default_plot,
here::here("figures", "raw", "rank-rank-scanpy-default-seurat-default.rds"))Scanpy Wilcox vs Seurat Wilcox
Disagreement caused (mainly) by tie-correction differences and also by regulation ranking differences. (Statistic vs p-value ranking has no effect.)
rank_rank_plot(
get_top_marker_genes(scanpy_wilcox, cluster = "B", n = 40),
get_top_marker_genes(seurat_wilcox, cluster = "B", n = 40),
label1 = "Scanpy Wilcoxon",
label2 = "Seurat Wilcoxon"
) +
ggtitle("Scanpy Wilcoxon vs Seurat Wilcoxon")![]()
Scanpy tie-corrected Wilcoxon vs Seurat Wilcox
Disagreement caused by regulation ranking difference (Smaller than for t-test due to enrichment for up-regulated genes.)
rank_rank_plot(
get_top_marker_genes(scanpy_wilcox_tc, cluster = "B", n = 40),
get_top_marker_genes(seurat_wilcox, cluster = "B", n = 40),
label1 = "Scanpy Wilcoxon (tie-corrected)",
label2 = "Seurat Wilcoxon"
) +
ggtitle("Scanpy Wilcoxon (tie-corrected) vs Seurat Wilcoxon")![]()
Scanpy tie-corrected Wilcoxon (ranking by absolute value) vs Seurat Wilcox
Complete agreement. (NB: Two p-values are 0 but the log fold change order is identical to the statistic order, see Zeisel, oligodendrocyte example below.)
rank_rank_plot(
get_top_marker_genes(scanpy_wilcox_tc_rankby_abs, cluster = "B", n = 40),
get_top_marker_genes(seurat_wilcox, cluster = "B", n = 40),
label1 = "Scanpy Wilcoxon (tie-corrected, ranking by absolute value)",
label2 = "Seurat Wilcoxon"
) +
ggtitle("Scanpy Wilcoxon (tie-corrected, ranking by absolute value) vs Seurat Wilcoxon")![]()
Scanpy t vs Seurat t
Disagreement caused mainly by regulation ranking difference, and issues with ranking by the t-statistic when using Welch’s t-test.
rank_rank_plot(
get_top_marker_genes(scanpy_t_raw, cluster = "B", n = 40),
get_top_marker_genes(seurat_t, cluster = "B", n = 40),
label1 = "Scanpy t",
label2 = "Seurat t"
) +
ggtitle("Scanpy t vs Seurat t")![]()
Scanpy t (ranking by absolute value) vs Seurat t
Disagreement caused by issues with ranking by the t-statistic when using Welch’s t-test.
rr_scanpy_t_abs_seurat_t <- rank_rank_plot(
get_top_marker_genes(scanpy_t_abs, cluster = "B", n = 20),
get_top_marker_genes(seurat_t, cluster = "B", n = 20),
label1 = "Scanpy t\n(ranking by absolute value)",
label2 = "Seurat t"
) +
theme(axis.ticks.y = element_blank())
rr_scanpy_t_abs_seurat_t![]()
saveRDS(
rr_scanpy_t_abs_seurat_t,
here::here("figures", "raw", "rr-scanpy-t-abs-seurat-t.rds")
)Scanpy tie-corrected Wilcoxon (ranking by absolute value) vs Seurat Wilcox (Zeisel data, oligodendrocyte cluster)
Disagreement caused solely difference in p-value vs statistic ranking. The oligodendrocyte cluster has a large number of zero p-values.
zeisel_seurat_wilcox <- readRDS(
here::here("results", "real_data", "zeisel-seurat_wilcox.rds")
)
zeisel_scanpy_wilcox_tc_abs <- readRDS(
here::here("results", "real_data",
"zeisel-scanpy_wilcoxontiecorrect_rankby_abs.rds")
)
n <- 40
cluster <- "oligodendrocytes"
rank_rank_plot(
get_top_marker_genes(zeisel_seurat_wilcox, cluster = cluster, n = n),
get_top_marker_genes(zeisel_scanpy_wilcox_tc_abs, cluster = cluster, n = n),
label1 = "Seurat Wilcoxon",
label2 = "Scanpy Wilcoxon (tie corrected, ranking by absolute value)"
) +
ggtitle("Scanpy Wilcoxon (tie-corrected, ranking by absolute value) vs Seurat Wilcoxon")![]()
Load simulated data
n_cells_data <- retrive_simulation_parameters() %>%
filter(sim_label == "num_cells") %>%
rowwise() %>%
mutate(
mgs_raw = list(readRDS(full_filename)$result),
mgs = list(split(mgs_raw, mgs_raw$cluster))
) %>%
ungroup() %>%
unnest_longer(
col = mgs,
values_to = "mgs",
indices_to = "cluster"
) %>%
select(-mgs_raw)Log fold-change concordance
n <- 20
logfc_mgs <- n_cells_data %>%
filter(cluster == "Group2", rep == 1) %>%
filter(pars %in% "logfc_raw") %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n))) %>%
ungroup() %>%
select(logfc_mgs = mgs, batchCells)
rest_mgs <- n_cells_data %>%
filter(cluster == "Group2", rep == 1) %>%
filter(method %in% c("seurat", "scanpy")) %>%
# We do not see the effect for ROC because it does not sort by p-value.
filter(pars != "seurat_roc") %>%
# The GLM use a differently calculated log-fc statistic.
filter(!(pars %in% c("seurat_negbinom", "seurat_poisson"))) %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n))) %>%
ungroup() %>%
dplyr::rename(other_mgs = mgs)
logfc_abs_prop_intersect_plot <- rest_mgs %>%
left_join(logfc_mgs, by = "batchCells") %>%
rowwise() %>%
mutate(
prop_intersect = length(intersect(logfc_mgs$gene, other_mgs$gene)) / n
) %>%
mutate(plot_method = method_lookup[method]) %>%
ungroup() %>%
ggplot(aes(x = factor(batchCells), y = prop_intersect,
fill = factor(plot_method))) +
geom_boxplot() +
coord_cartesian(ylim = c(0.5, 1)) +
labs(
x = "Total cells simulated",
y = "Intersection with raw log fold-change method",
fill = "Package"
) +
scale_fill_manual(values = c("#1e90ff", "#ff0000")) +
theme_bw()
logfc_abs_prop_intersect_plot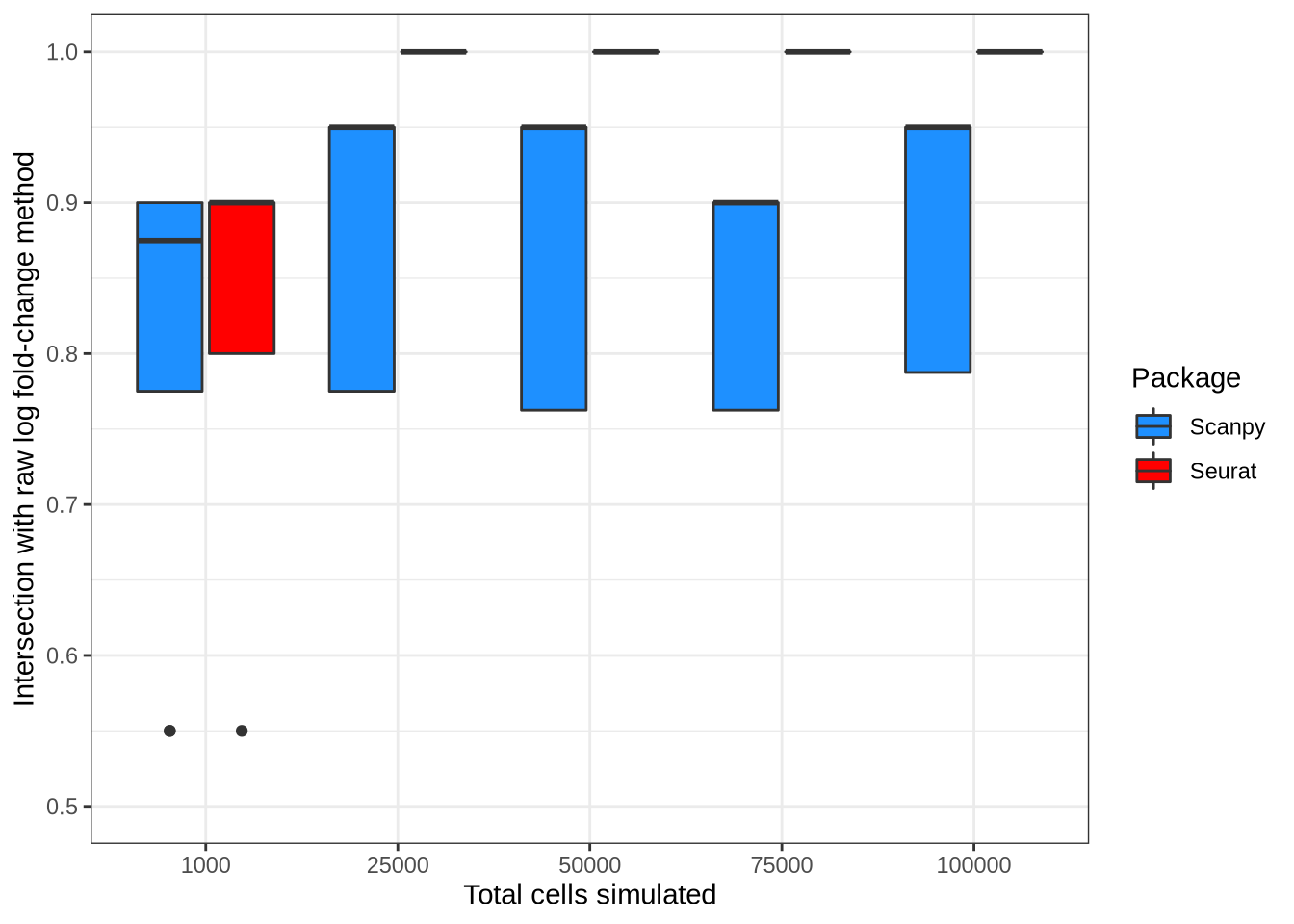
saveRDS(
logfc_abs_prop_intersect_plot,
here::here("figures", "raw", "logfc-abs-prop-intersect.rds")
)plot_dendrogram_by_dataset <- function(data, data_id) {
n <- 10
all_concordance_data <- data %>%
filter(data_id == !!data_id) %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = n)$gene)) %>%
ungroup() %>%
select(pars, method, cluster, data_id, mgs)
intersection_data <- all_concordance_data %>%
expand_grid(pars_2 = unique(all_concordance_data$pars)) %>%
left_join(
dplyr::rename(all_concordance_data, mgs_2 = mgs),
by = c("pars_2" = "pars", "cluster")
) %>%
rowwise() %>%
mutate(prop_intersect = length(intersect(mgs, mgs_2)) / n) %>%
ungroup() %>%
select(pars, pars_2, cluster, prop_intersect) %>%
group_by(pars, pars_2) %>%
summarise(prop_intersect = mean(prop_intersect), .groups = "drop") %>%
pivot_wider(names_from = pars, values_from = prop_intersect)
intersection_mat <- as.matrix(intersection_data[, -1])
rownames(intersection_mat) <- intersection_data[[1]]
hier_clust <- hclust(dist(intersection_mat))
hier_clust$labels <- pars_lookup[hier_clust$labels]
hier_clust_data <- dendro_data(hier_clust, type = "rectangle")
dendrogram_plot <- ggplot() +
geom_segment(data = segment(hier_clust_data),
aes(x = x, y = y, xend = xend, yend = yend)
) +
geom_text(data = label(hier_clust_data),
aes(x = x, y = y, label = label, hjust = 0),
size = 4
) +
coord_flip(xlim = c(3, 57)) +
scale_y_reverse(limits = c(4, -1.5)) +
theme_dendro()
dendrogram_plot
}
pbmc3k_dendro <- plot_dendrogram_by_dataset(concordance_data, "pbmc3k") +
ggtitle("pbmc3k")
endothelial_dendro <- plot_dendrogram_by_dataset(concordance_data, "endothelial") +
ggtitle("Endothelial")
ren_dendro <- plot_dendrogram_by_dataset(concordance_data, "ren") +
ggtitle("Ren")
astrocyte_dendro <- plot_dendrogram_by_dataset(concordance_data, "astrocyte") +
ggtitle("Astrocyte")
dataset_dendro <- pbmc3k_dendro + endothelial_dendro + ren_dendro +
astrocyte_dendro +
plot_layout(guides = "collect") +
plot_annotation(tag_levels = "a") &
theme(plot.tag = element_text(size = 18))
ggsave(
filename = here::here("figures", "final", "dataset-dendro.pdf"),
plot = dataset_dendro,
width = 14,
height = 18,
units = "in"
)n_cells_data <- lapply(
list.files(here::here("data", "real_data"), full.names = TRUE),
function(x) {
data <- readRDS(x)
out <- tibble(
data_id = tools::file_path_sans_ext(basename(x)),
cell_type = unique(data$label),
n = as.vector(table(data$label))
)
rm(data)
out
}
) %>%
bind_rows(!!!.)n <- 10
all_concordance_data <- concordance_data %>%
rowwise() %>%
mutate(mgs = list(get_top_sel_mgs(mgs, n = n)$gene)) %>%
ungroup() %>%
select(pars, method, cluster, data_id, mgs)
intersect_n_cells_data <- all_concordance_data %>%
expand_grid(pars_2 = unique(all_concordance_data$pars)) %>%
left_join(
dplyr::rename(all_concordance_data, mgs_2 = mgs),
by = c("pars_2" = "pars", "cluster")
) %>%
rowwise() %>%
mutate(prop_intersect = length(intersect(mgs, mgs_2)) / n) %>%
ungroup() %>%
select(pars, pars_2, cluster, prop_intersect, data_id.x) %>%
group_by(cluster, data_id.x) %>%
summarise(prop_intersect = mean(prop_intersect), .groups = "drop") %>%
left_join(n_cells_data, by = c("cluster" = "cell_type", "data_id.x" = "data_id"))
intersect_n_cells_plot <- intersect_n_cells_data %>%
filter(!is.na(n)) %>%
ggplot(aes(n, prop_intersect)) +
geom_point() +
labs(
x = "Number of cells",
y = "Intersect proportion"
) +
theme_bw()
ggsave(
filename = here::here("figures", "final", "intersect-n-cells.pdf"),
plot = intersect_n_cells_plot ,
width = 8,
height = 8,
units = "in"
)
devtools::session_info()─ Session info ──────────────────────────────────────────────────────────────
hash: person frowning: dark skin tone, elevator, right-facing fist: medium-dark skin tone
setting value
version R version 4.1.2 (2021-11-01)
os Red Hat Enterprise Linux 9.2 (Plow)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Melbourne
date 2024-01-01
pandoc 2.18 @ /apps/easybuild-2022/easybuild/software/MPI/GCC/11.3.0/OpenMPI/4.1.4/RStudio-Server/2022.07.2+576-Java-11-R-4.1.2/bin/pandoc/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
assertthat 0.2.1 2019-03-21 [2] CRAN (R 4.1.2)
beachmat 2.10.0 2021-10-26 [1] Bioconductor
beeswarm 0.4.0 2021-06-01 [2] CRAN (R 4.1.2)
Biobase * 2.54.0 2021-10-26 [1] Bioconductor
BiocGenerics * 0.40.0 2021-10-26 [1] Bioconductor
BiocNeighbors 1.12.0 2021-10-26 [1] Bioconductor
BiocParallel 1.28.3 2021-12-09 [1] Bioconductor
BiocSingular 1.10.0 2021-10-26 [1] Bioconductor
bitops 1.0-7 2021-04-24 [2] CRAN (R 4.1.2)
bslib 0.3.1 2021-10-06 [1] CRAN (R 4.1.0)
cachem 1.0.6 2021-08-19 [1] CRAN (R 4.1.0)
callr 3.7.0 2021-04-20 [2] CRAN (R 4.1.2)
cli 3.6.1 2023-03-23 [1] CRAN (R 4.1.0)
colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.1.0)
crayon 1.5.1 2022-03-26 [1] CRAN (R 4.1.0)
DBI 1.1.2 2021-12-20 [1] CRAN (R 4.1.0)
DelayedArray 0.20.0 2021-10-26 [1] Bioconductor
DelayedMatrixStats 1.16.0 2021-10-26 [1] Bioconductor
desc 1.4.0 2021-09-28 [2] CRAN (R 4.1.2)
devtools 2.4.2 2021-06-07 [2] CRAN (R 4.1.2)
dichromat 2.0-0 2013-01-24 [2] CRAN (R 4.1.2)
digest 0.6.29 2021-12-01 [1] CRAN (R 4.1.0)
dplyr * 1.0.9 2022-04-28 [1] CRAN (R 4.1.0)
ellipsis 0.3.2 2021-04-29 [2] CRAN (R 4.1.2)
evaluate 0.14 2019-05-28 [2] CRAN (R 4.1.2)
fansi 1.0.4 2023-01-22 [1] CRAN (R 4.1.0)
farver 2.1.1 2022-07-06 [1] CRAN (R 4.1.0)
fastmap 1.1.0 2021-01-25 [2] CRAN (R 4.1.2)
forcats * 0.5.1 2021-01-27 [2] CRAN (R 4.1.2)
fs 1.5.2 2021-12-08 [1] CRAN (R 4.1.0)
generics 0.1.3 2022-07-05 [1] CRAN (R 4.1.0)
GenomeInfoDb * 1.30.0 2021-10-26 [1] Bioconductor
GenomeInfoDbData 1.2.7 2021-12-03 [1] Bioconductor
GenomicRanges * 1.46.1 2021-11-18 [1] Bioconductor
ggbeeswarm 0.6.0 2017-08-07 [2] CRAN (R 4.1.2)
ggdendro * 0.1.23 2022-02-16 [1] CRAN (R 4.1.0)
ggplot2 * 3.3.6 2022-05-03 [1] CRAN (R 4.1.0)
ggrepel * 0.9.1 2021-01-15 [2] CRAN (R 4.1.2)
ggupset * 0.3.0 2020-05-05 [1] CRAN (R 4.1.0)
git2r 0.28.0 2021-01-10 [2] CRAN (R 4.1.2)
glue 1.6.0 2021-12-17 [1] CRAN (R 4.1.0)
gridExtra 2.3 2017-09-09 [2] CRAN (R 4.1.2)
gtable 0.3.0 2019-03-25 [2] CRAN (R 4.1.2)
here 1.0.1 2020-12-13 [1] CRAN (R 4.1.0)
highr 0.9 2021-04-16 [2] CRAN (R 4.1.2)
htmltools 0.5.2 2021-08-25 [1] CRAN (R 4.1.0)
httpuv 1.6.5 2022-01-05 [1] CRAN (R 4.1.0)
IRanges * 2.28.0 2021-10-26 [1] Bioconductor
irlba 2.3.5 2021-12-06 [1] CRAN (R 4.1.0)
jquerylib 0.1.4 2021-04-26 [2] CRAN (R 4.1.2)
jsonlite 1.8.0 2022-02-22 [1] CRAN (R 4.1.0)
knitr 1.36 2021-09-29 [1] CRAN (R 4.1.0)
labeling 0.4.2 2020-10-20 [2] CRAN (R 4.1.2)
later 1.3.0 2021-08-18 [1] CRAN (R 4.1.0)
lattice 0.20-45 2021-09-22 [2] CRAN (R 4.1.2)
lifecycle 1.0.1 2021-09-24 [1] CRAN (R 4.1.0)
logisticPCA * 0.2 2016-03-14 [1] CRAN (R 4.1.0)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.1.0)
mapproj 1.2.7 2020-02-03 [1] CRAN (R 4.1.0)
maps 3.4.0 2021-09-25 [2] CRAN (R 4.1.2)
MASS 7.3-54 2021-05-03 [2] CRAN (R 4.1.2)
Matrix 1.3-4 2021-06-01 [2] CRAN (R 4.1.2)
MatrixGenerics * 1.6.0 2021-10-26 [1] Bioconductor
matrixStats * 0.62.0 2022-04-19 [1] CRAN (R 4.1.0)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.1.0)
munsell 0.5.0 2018-06-12 [2] CRAN (R 4.1.2)
pals * 1.7 2021-04-17 [1] CRAN (R 4.1.0)
patchwork * 1.1.1 2020-12-17 [2] CRAN (R 4.1.2)
pillar 1.7.0 2022-02-01 [1] CRAN (R 4.1.0)
pkgbuild 1.2.0 2020-12-15 [2] CRAN (R 4.1.2)
pkgconfig 2.0.3 2019-09-22 [2] CRAN (R 4.1.2)
pkgload 1.2.3 2021-10-13 [2] CRAN (R 4.1.2)
prettyunits 1.1.1 2020-01-24 [2] CRAN (R 4.1.2)
processx 3.5.2 2021-04-30 [2] CRAN (R 4.1.2)
promises 1.2.0.1 2021-02-11 [2] CRAN (R 4.1.2)
ps 1.7.1 2022-06-18 [1] CRAN (R 4.1.0)
purrr * 0.3.4 2020-04-17 [2] CRAN (R 4.1.2)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.1.0)
Rcpp 1.0.8.3 2022-03-17 [1] CRAN (R 4.1.0)
RCurl 1.98-1.5 2021-09-17 [1] CRAN (R 4.1.0)
remotes 2.4.2 2021-11-30 [1] CRAN (R 4.1.0)
rlang 1.0.3 2022-06-27 [1] CRAN (R 4.1.0)
rmarkdown 2.14 2022-04-25 [1] CRAN (R 4.1.0)
rprojroot 2.0.3 2022-04-02 [1] CRAN (R 4.1.0)
rstudioapi 0.14 2022-08-22 [1] CRAN (R 4.1.0)
rsvd 1.0.5 2021-04-16 [1] CRAN (R 4.1.0)
S4Vectors * 0.32.3 2021-11-21 [1] Bioconductor
sass 0.4.1 2022-03-23 [1] CRAN (R 4.1.0)
ScaledMatrix 1.2.0 2021-10-26 [1] Bioconductor
scales 1.2.1 2022-08-20 [1] CRAN (R 4.1.0)
scater * 1.22.0 2021-10-26 [1] Bioconductor
scuttle * 1.4.0 2021-10-26 [1] Bioconductor
sessioninfo 1.2.0 2021-10-31 [2] CRAN (R 4.1.2)
SingleCellExperiment * 1.16.0 2021-10-26 [1] Bioconductor
sparseMatrixStats 1.6.0 2021-10-26 [1] Bioconductor
stringi 1.7.6 2021-11-29 [1] CRAN (R 4.1.0)
stringr 1.4.0 2019-02-10 [2] CRAN (R 4.1.2)
SummarizedExperiment * 1.24.0 2021-10-26 [1] Bioconductor
testthat 3.1.0 2021-10-04 [2] CRAN (R 4.1.2)
tibble * 3.1.7 2022-05-03 [1] CRAN (R 4.1.0)
tidyr * 1.2.0 2022-02-01 [1] CRAN (R 4.1.0)
tidyselect 1.1.2 2022-02-21 [1] CRAN (R 4.1.0)
topconfects * 1.10.0 2021-10-26 [1] Bioconductor
usethis 2.1.3 2021-10-27 [2] CRAN (R 4.1.2)
utf8 1.2.3 2023-01-31 [1] CRAN (R 4.1.0)
vctrs 0.4.1 2022-04-13 [1] CRAN (R 4.1.0)
vipor 0.4.5 2017-03-22 [2] CRAN (R 4.1.2)
viridis 0.6.2 2021-10-13 [1] CRAN (R 4.1.0)
viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.1.0)
whisker 0.4 2019-08-28 [2] CRAN (R 4.1.2)
withr 2.5.0 2022-03-03 [1] CRAN (R 4.1.0)
workflowr 1.7.0 2021-12-21 [1] CRAN (R 4.1.0)
xfun 0.31 2022-05-10 [1] CRAN (R 4.1.0)
XVector 0.34.0 2021-10-26 [1] Bioconductor
yaml 2.3.5 2022-02-21 [1] CRAN (R 4.1.0)
zlibbioc 1.40.0 2021-10-26 [1] Bioconductor
[1] /home/jpullin/R/x86_64-pc-linux-gnu-library/4.1
[2] /apps/easybuild-2022/easybuild/software/MPI/GCC/11.3.0/OpenMPI/4.1.4/R/4.1.2/lib64/R/library
──────────────────────────────────────────────────────────────────────────────