17 “Ideal” scRNAseq pipeline (as of Oct 2019)
17.1 Experimental Design
- Avoid confounding biological and batch effects (Figure 17.1)
- Multiple conditions should be captured on the same chip if possible
- Perform multiple replicates of each condition where replicates of different conditions should be performed together if possible
- Statistics cannot correct a completely confounded experiment!
- Unique molecular identifiers
- Greatly reduce noise in data
- May reduce gene detection rates (unclear if it is UMIs or other protocol differences)
- Lose splicing information
- Use longer UMIs (~10bp)
- Correct for sequencing errors in UMIs using UMI-tools
- Spike-ins
- Useful for quality control
- May be useful for normalizing read counts
- Can be used to approximate cell-size/RNA content (if relevant to biological question)
- Often exhibit higher noise than endogenous genes (pipetting errors, mixture quality)
- Requires more sequencing to get enough endogenous reads per cell
- Cell number vs Read depth
- Gene detection plateaus starting from 1 million reads per cell
- Transcription factor detection (regulatory networks) require high read depth and most sensitive protocols (i.e. Fluidigm C1)
- Cell clustering & cell-type identification benefits from large number of cells and doesn’t requireas high sequencing depth (~100,000 reads per cell).
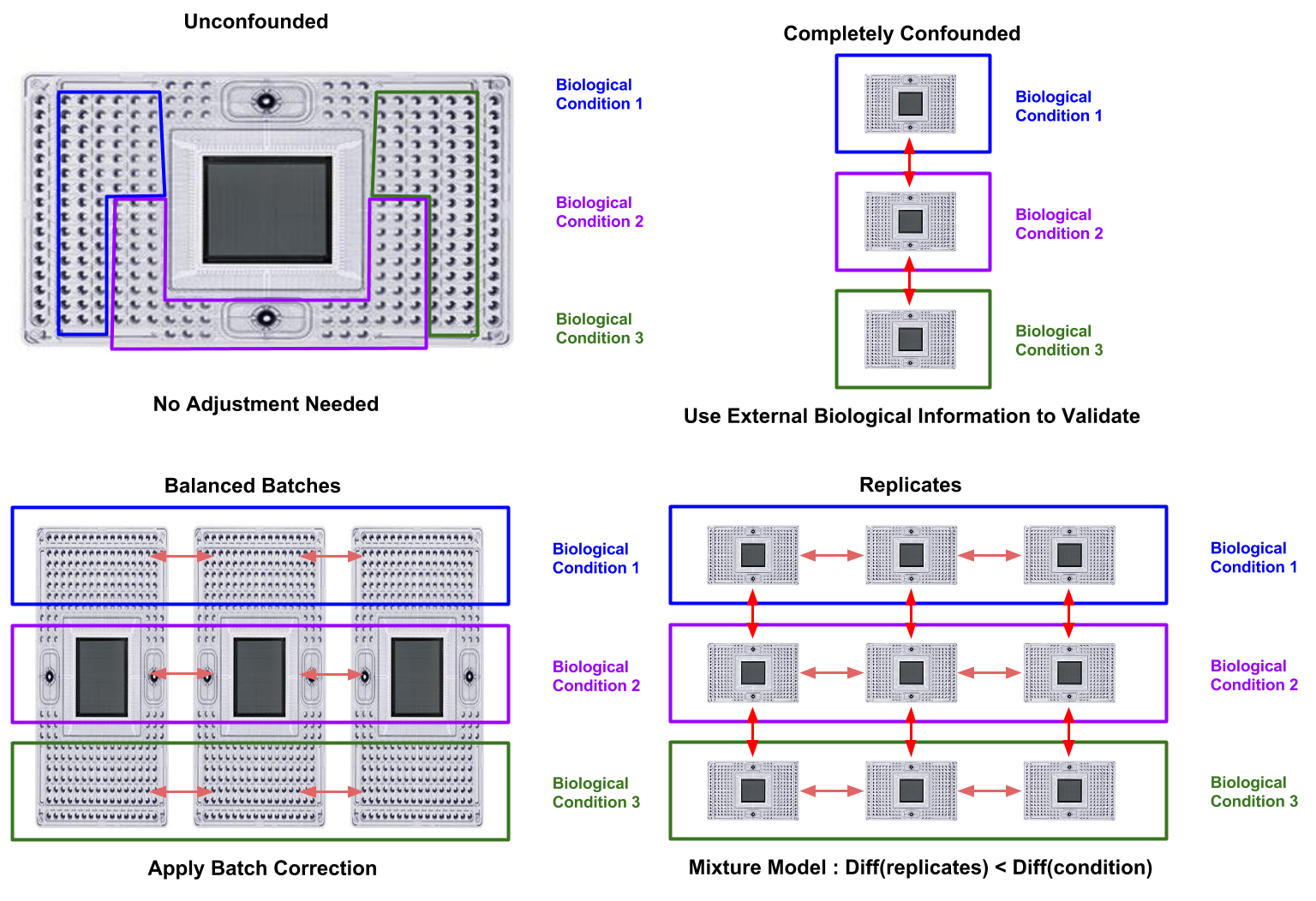
Figure 17.1: Appropriate approaches to batch effects in scRNASeq. Red arrows indicate batch effects which are (pale) or are not (vibrant) correctable through batch-correction.
17.2 Processing Reads
- Read QC & Trimming
- Mapping
- Quantification
- Small dataset, no UMIs : featureCounts
- Large datasets, no UMIs: Salmon, kallisto
- UMI dataset : UMI-tools + featureCounts
17.3 Preparing Expression Matrix
- Cell QC
- scater
- consider: mtRNA, rRNA, spike-ins (if available), number of detected genes per cell, total reads/molecules per cell
- Library Size Normalization
- Batch correction (if appropriate)
- Replicates/Confounded RUVs
- Unknown or unbalanced biological groups mnnCorrect
- Balanced design ComBat
17.4 Biological Interpretation
- Feature Selection
- Clustering and Marker Gene Identification
- Pseudotime
- Differential Expression