13 Imputation
library(scImpute)
library(SC3)
library(scater)
library(SingleCellExperiment)
library(mclust)
library(DrImpute)
set.seed(1234567)As discussed previously, one of the main challenges when analyzing scRNA-seq data is the presence of zeros, or dropouts. The dropouts are assumed to have arisen for three possible reasons:
- The gene was not expressed in the cell and hence there are no transcripts to sequence
- The gene was expressed, but for some reason the transcripts were lost somewhere prior to sequencing
- The gene was expressed and transcripts were captured and turned into cDNA, but the sequencing depth was not sufficient to produce any reads.
Thus, dropouts could be result of experimental shortcomings, and if this is the case then we would like to provide computational corrections. One possible solution is to impute the dropouts in the expression matrix. To be able to impute gene expression values, one must have an underlying model. However, since we do not know which dropout events are technical artefacts and which correspond to the transcript being truly absent, imputation is a difficult challenge and prone to creating false-positive results in downstream analysis.
There are many different imputation methods available we will consider three fast, published methods: DrImpute and scImpute (Li and Li 2017).
DrImpute and scImpute both use a model to determine which zeros are technical and impute only those values. Both use clustering to identify a group of cells that are assumed to have homogenous expression. DrImpute imputes all values that are not consistently zero in all cells of a cluster. Whereas, scImpute uses a zero-inflated normal distribution fit to log-normalized expression values and imputed all inflated zeros.
13.0.1 scImpute
To test scImpute, we use the default parameters and we apply it to the Deng
dataset that we have worked with before. scImpute takes a .csv or .txt file as
an input:
deng <- readRDS("data/deng/deng-reads.rds")
write.csv(counts(deng), "deng.csv")
scimpute(
count_path = "deng.csv",
infile = "csv",
outfile = "txt",
out_dir = "./",
Kcluster = 10,
ncores = 2
)## [1] "reading in raw count matrix ..."
## [1] "number of genes in raw count matrix 22431"
## [1] "number of cells in raw count matrix 268"
## [1] "reading finished!"
## [1] "imputation starts ..."
## [1] "searching candidate neighbors ... "
## [1] "inferring cell similarities ..."
## [1] "dimension reduction ..."
## [1] "calculating cell distances ..."
## [1] "cluster sizes:"
## [1] 84 9 72 5 8 12 8 22 24 14
## [1] "estimating dropout probability for type 1 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 1 ..."
## [1] "estimating dropout probability for type 2 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 2 ..."
## [1] "estimating dropout probability for type 3 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 3 ..."
## [1] "estimating dropout probability for type 4 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 4 ..."
## [1] "estimating dropout probability for type 5 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 5 ..."
## [1] "estimating dropout probability for type 6 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 6 ..."
## [1] "estimating dropout probability for type 7 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 7 ..."
## [1] "estimating dropout probability for type 8 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 8 ..."
## [1] "estimating dropout probability for type 9 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 9 ..."
## [1] "estimating dropout probability for type 10 ..."
## [1] "searching for valid genes ..."
## [1] "imputing dropout values for type 10 ..."
## [1] "writing imputed count matrix ..."## [1] 17 18 88 111 126 177 186 229 244 247Now we can compare the results with original data by considering a PCA plot
res <- read.table("scimpute_count.txt")
colnames(res) <- NULL
res <- SingleCellExperiment(
assays = list(logcounts = log2(as.matrix(res) + 1)),
colData = colData(deng)
)
rowData(res)$feature_symbol <- rowData(deng)$feature_symbol
plotPCA(
res,
colour_by = "cell_type2"
)
Compare this result to the original data in Chapter 10.2. What are the most significant differences?
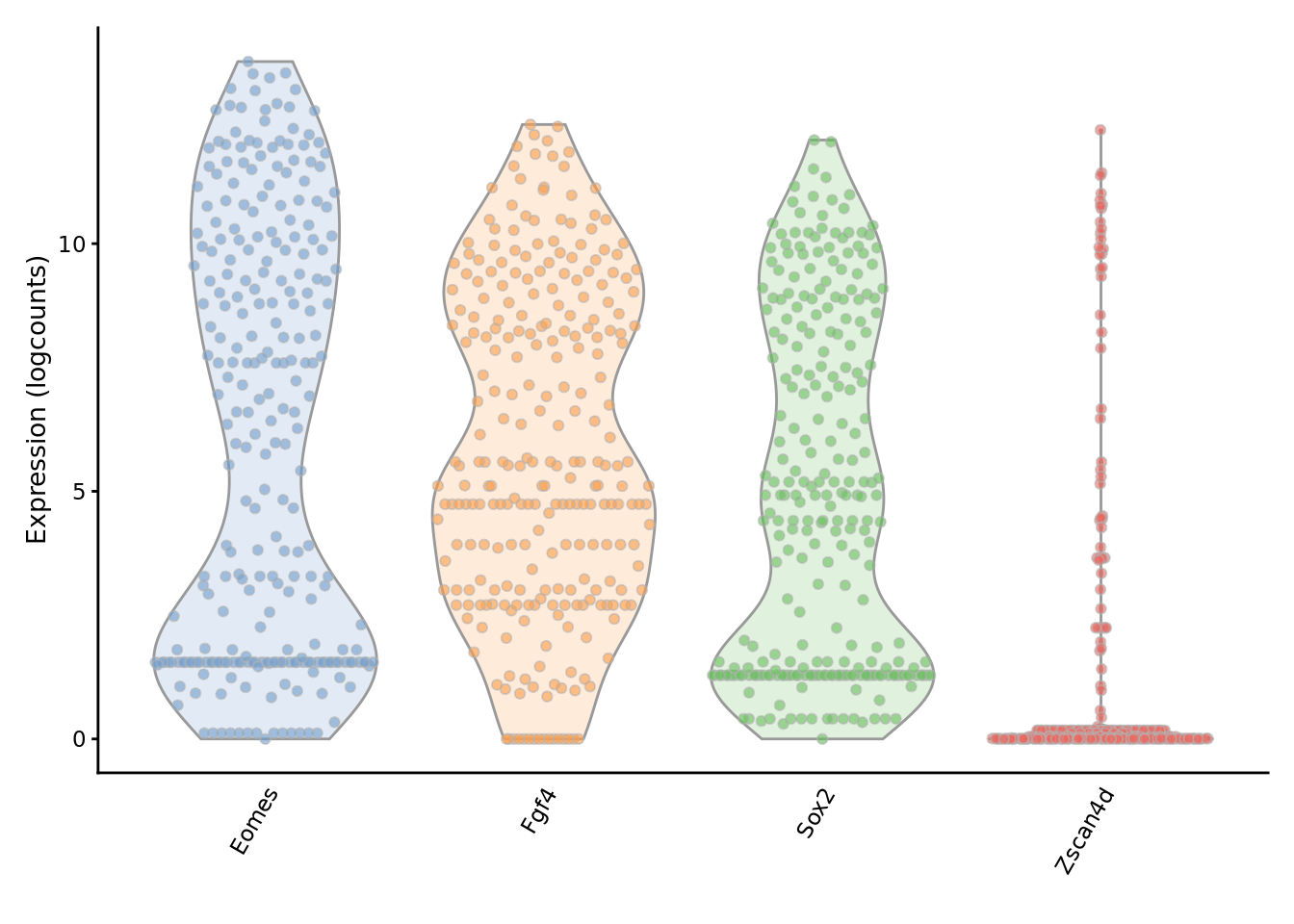
We can examine the expression of specific genes to directly see the effect of imputation on the expression distribution.
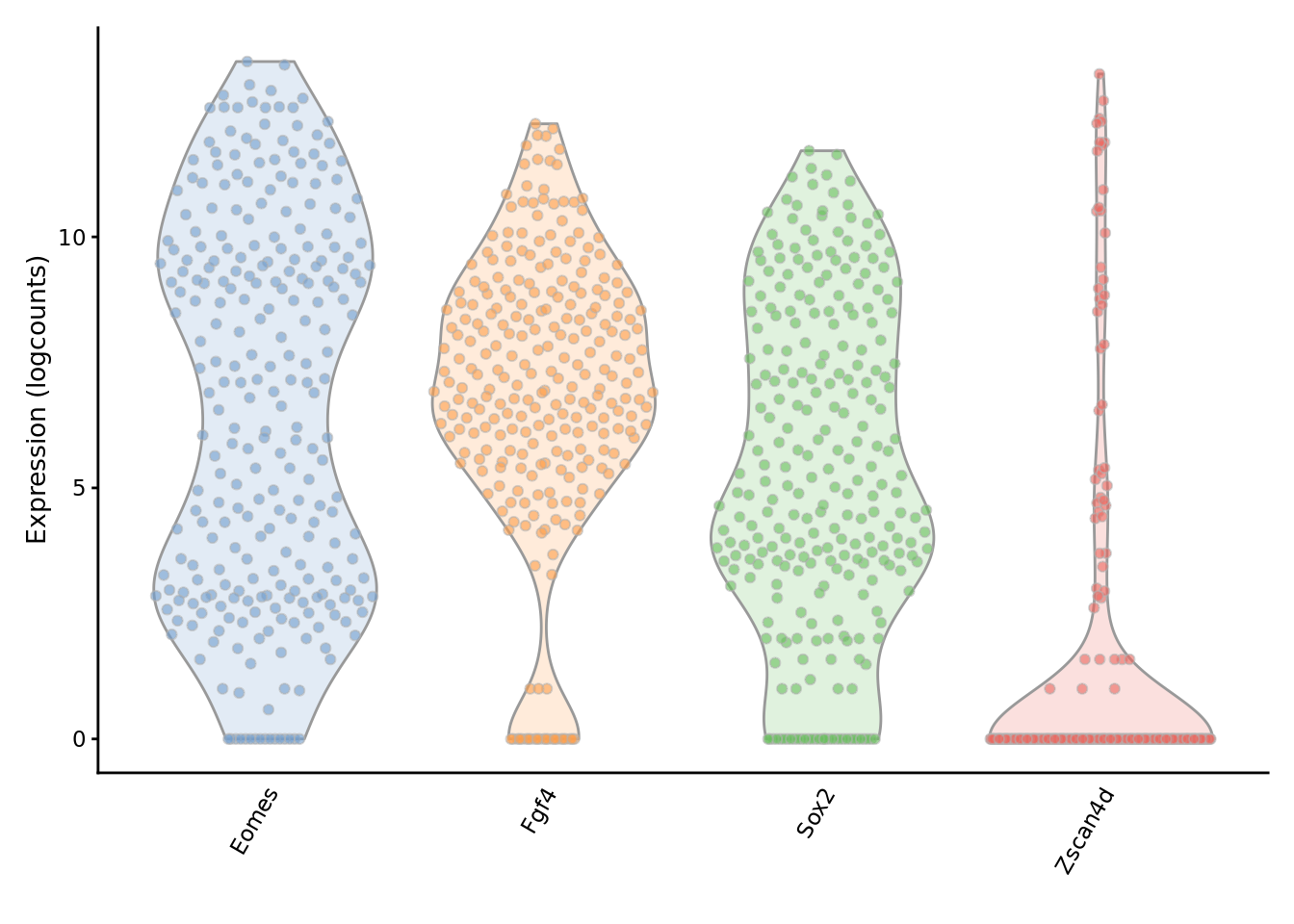
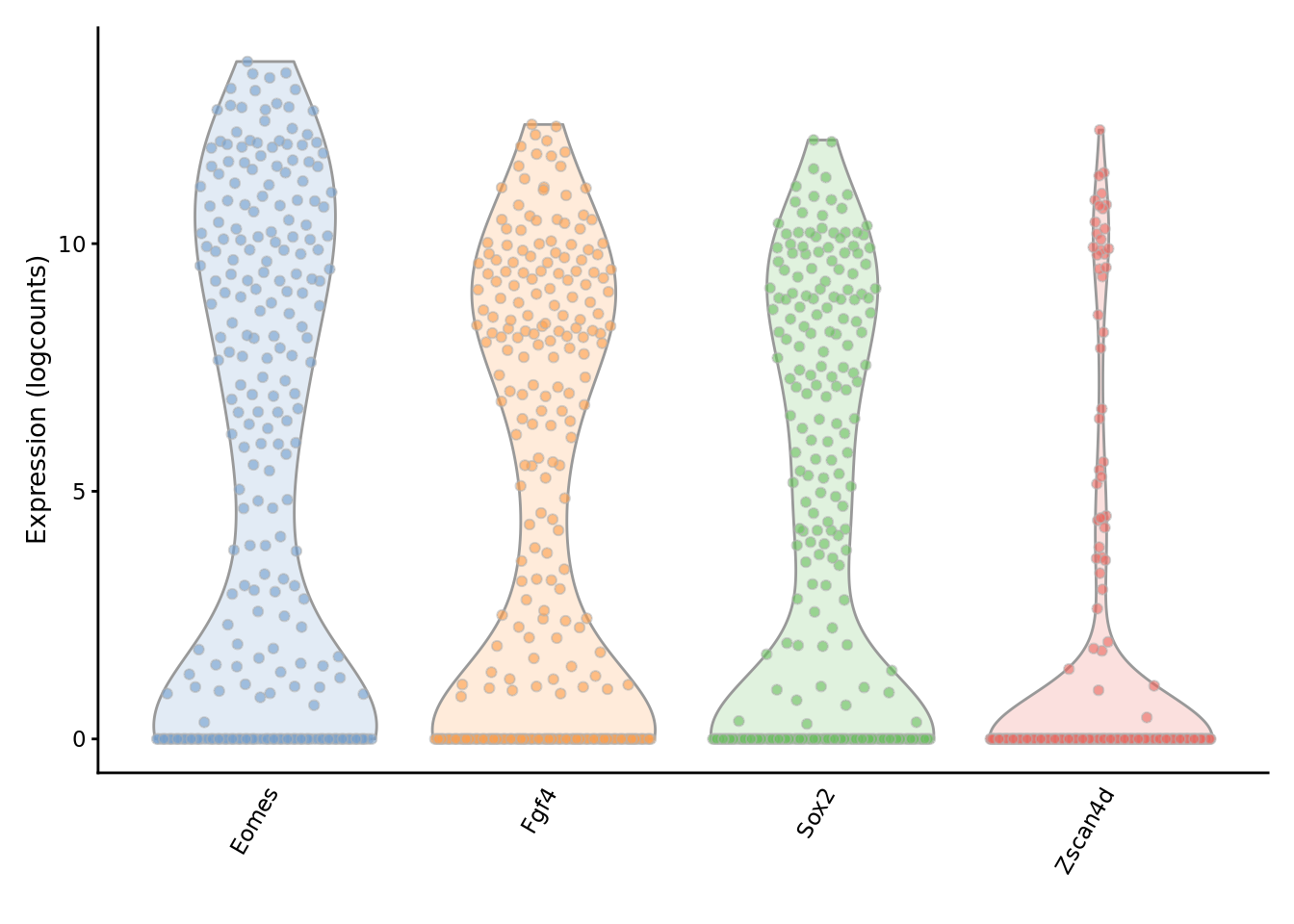
To evaluate the impact of the imputation, we use SC3 to cluster the imputed matrix
## [1] 6## [1] 0.5062983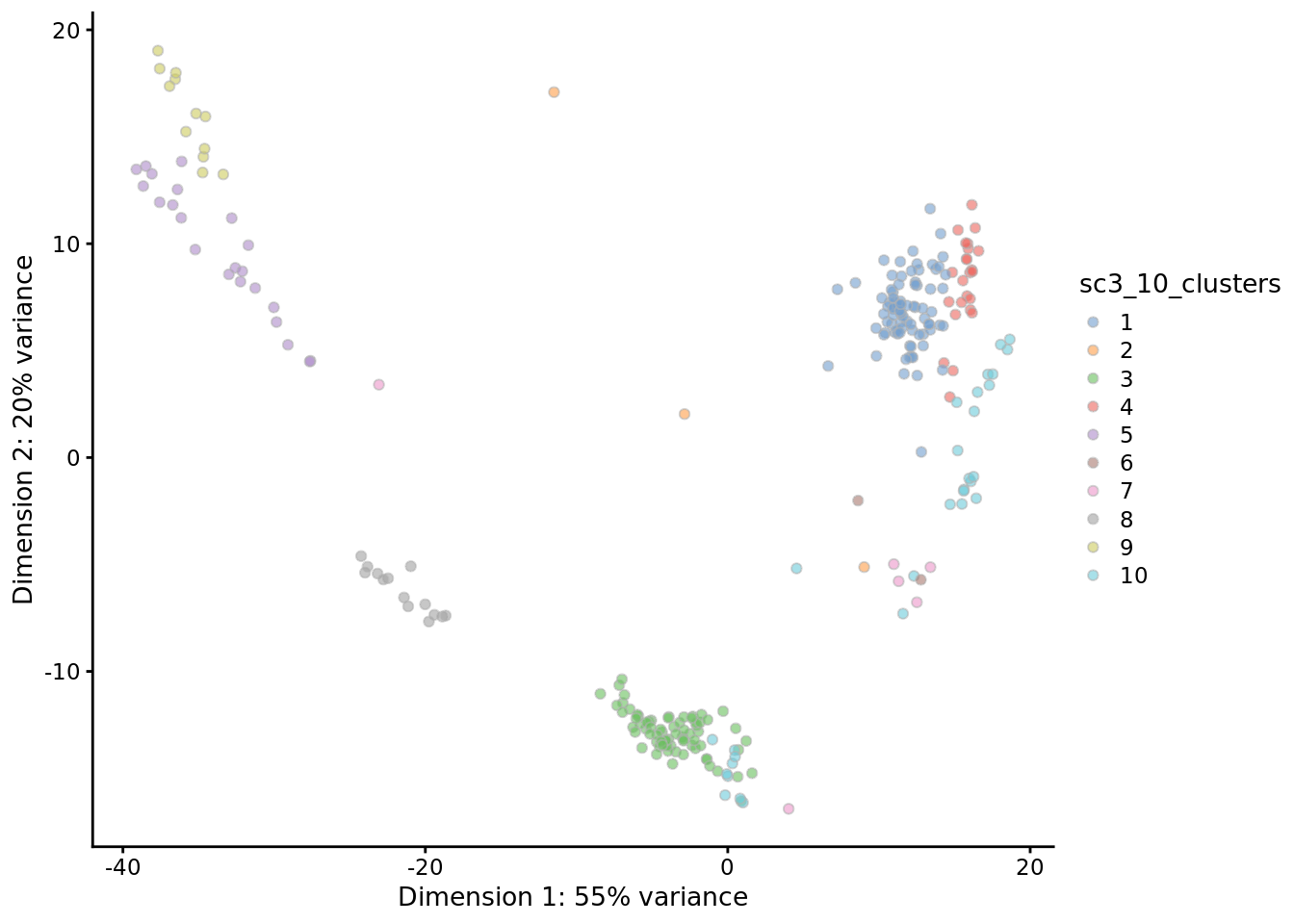
Exercise: Based on the PCA and the clustering results, do you think that imputation using scImpute is a good idea for the Deng dataset?
13.0.2 DrImpute
We can do the same for DrImpute. DrImpute runs on a log-normalized expression matrix directly in R, we generate this matrix using scater, then run DrImpute. Unlike scImpute, DrImpute considers the consensus imputation across a range of ks using two differ correlation distances:
## Calculating Spearman distance.
## Calculating Pearson distance.
## Clustering for k : 8
## Clustering for k : 9
## Clustering for k : 10
## Clustering for k : 11
## Clustering for k : 12
## cls object have 10 number of clustering sets.
##
##
## Zero percentage :
## Before impute : 60 percent.
## After impute : 17 percent.
## 43 percent of zeros are imputed.colnames(res) <- colnames(deng)
rownames(res) <- rownames(deng)
res <- SingleCellExperiment(
assays = list(logcounts = as.matrix(res)),
colData = colData(deng)
)
rowData(res)$feature_symbol <- rowData(deng)$feature_symbol
plotPCA(
res,
colour_by = "cell_type2"
)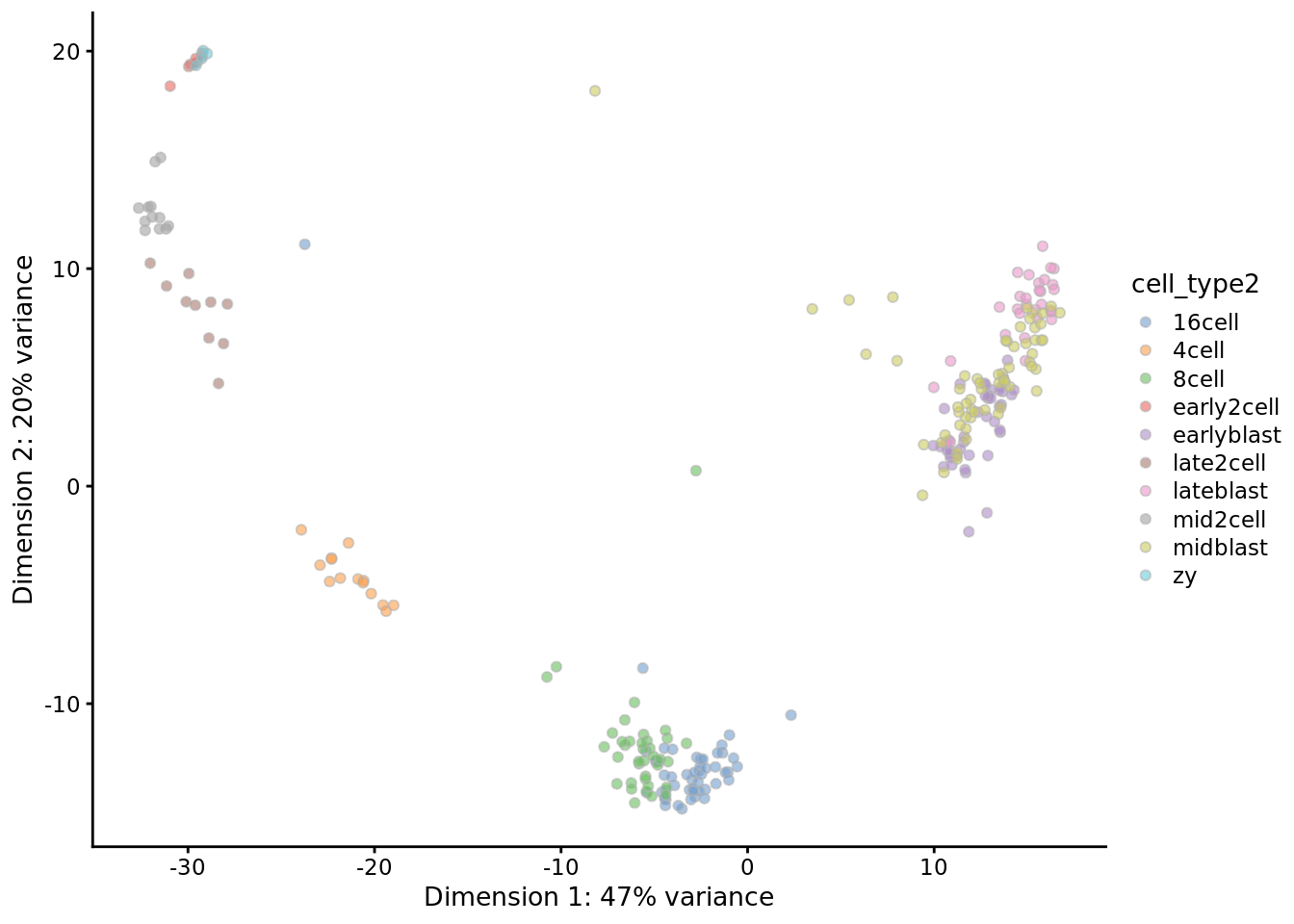
Exercise: Check the sc3 clustering of the DrImpute matrix, do you think that
imputation using DrImpute is a good idea for the Deng dataset?
Exercise: What is the difference between scImpute and DrImpute based on
the PCA and clustering analysis? Which one do you think is best to use?
13.0.3 sessionInfo()
## R version 3.6.0 (2019-04-26)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
##
## locale:
## [1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
## [5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
## [7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats4 parallel stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] DrImpute_1.0 mclust_5.4.5
## [3] scater_1.12.2 ggplot2_3.2.1
## [5] SingleCellExperiment_1.6.0 SummarizedExperiment_1.14.1
## [7] DelayedArray_0.10.0 BiocParallel_1.18.1
## [9] matrixStats_0.55.0 Biobase_2.44.0
## [11] GenomicRanges_1.36.1 GenomeInfoDb_1.20.0
## [13] IRanges_2.18.3 S4Vectors_0.22.1
## [15] BiocGenerics_0.30.0 SC3_1.12.0
## [17] scImpute_0.0.9 doParallel_1.0.15
## [19] iterators_1.0.12 foreach_1.4.7
## [21] penalized_0.9-51 survival_2.43-3
##
## loaded via a namespace (and not attached):
## [1] bitops_1.0-6 RColorBrewer_1.1-2
## [3] tools_3.6.0 doRNG_1.7.1
## [5] irlba_2.3.3 R6_2.4.0
## [7] vipor_0.4.5 KernSmooth_2.23-15
## [9] lazyeval_0.2.2 colorspace_1.4-1
## [11] withr_2.1.2 gridExtra_2.3
## [13] tidyselect_0.2.5 compiler_3.6.0
## [15] BiocNeighbors_1.2.0 pkgmaker_0.27
## [17] labeling_0.3 bookdown_0.13
## [19] caTools_1.17.1.2 scales_1.0.0
## [21] DEoptimR_1.0-8 mvtnorm_1.0-11
## [23] robustbase_0.93-5 stringr_1.4.0
## [25] digest_0.6.21 rmarkdown_1.15
## [27] XVector_0.24.0 rrcov_1.4-7
## [29] pkgconfig_2.0.3 htmltools_0.3.6
## [31] bibtex_0.4.2 WriteXLS_5.0.0
## [33] rlang_0.4.0 DelayedMatrixStats_1.6.1
## [35] shiny_1.3.2 gtools_3.8.1
## [37] dplyr_0.8.3 BiocSingular_1.0.0
## [39] RCurl_1.95-4.12 magrittr_1.5
## [41] GenomeInfoDbData_1.2.1 Matrix_1.2-17
## [43] ggbeeswarm_0.6.0 Rcpp_1.0.2
## [45] munsell_0.5.0 viridis_0.5.1
## [47] stringi_1.4.3 yaml_2.2.0
## [49] zlibbioc_1.30.0 gplots_3.0.1.1
## [51] grid_3.6.0 gdata_2.18.0
## [53] promises_1.0.1 crayon_1.3.4
## [55] lattice_0.20-38 cowplot_1.0.0
## [57] splines_3.6.0 knitr_1.25
## [59] pillar_1.4.2 rngtools_1.4
## [61] codetools_0.2-16 glue_1.3.1
## [63] evaluate_0.14 httpuv_1.5.2
## [65] gtable_0.3.0 purrr_0.3.2
## [67] kernlab_0.9-27 assertthat_0.2.1
## [69] xfun_0.9 mime_0.7
## [71] rsvd_1.0.2 xtable_1.8-4
## [73] e1071_1.7-2 later_0.8.0
## [75] viridisLite_0.3.0 class_7.3-15
## [77] pcaPP_1.9-73 tibble_2.1.3
## [79] pheatmap_1.0.12 beeswarm_0.2.3
## [81] registry_0.5-1 cluster_2.1.0
## [83] ROCR_1.0-7References
Li, Wei Vivian, and Jingyi Jessica Li. 2017. “scImpute: Accurate and Robust Imputation for Single Cell RNA-Seq Data.” bioRxiv, June, 141598.